承認日:2015年06月05日
改訂日:2015年12月23日
2016年02月09日(木
2017年2月24日(木
2018年1月24日(木
XX/XX/201X
アビラテロン酢酸塩錠の使用説明書
説明書をよく読み.医師の指導のもとでご使用ください。
1.薬剤名
について
一般名:アビラテロン酢酸塩錠
商品名:ゼコール®ザイティガ®。
英語名:Abiraterone Acetate Tablets。
Hanyu Pinyin: Cusuan Abitelong Pian
②【成分】
。
<主な成分: 酢酸アビラテロン
<化学名: 17-(3-ピリジル)-アンドロスト-5,16-ジエン-3β-アセテート
化学構造式: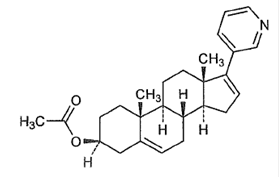
分子式:C26H33NO2
分子量: 391.55
賦形剤: 乳糖一水和物.架橋カルボキシメチルセルロースナトリウム.ポビドン(K29/K32).ドデシル硫酸ナトリウム.微結晶セルロース.コロイド状含水二酸化ケイ素.ステアリン酸マグネシウム
3. [プロパティ]本製品は白色またはオフホワイトの錠剤です。
4. 【適応症】本製品は.以下の治療にプレドニゾンまたはプレドニゾロンと併用して使用されます。
転移性デスモイド抵抗性前立腺癌(mCRPC)
新規に診断された高リスクの転移性治療感受性前立腺癌(mHSPC)(内分泌療法を受けていない.または内分泌療法を受けてから最長3ヶ月経過している者を含む)。
5. [仕様]250mg
6.用法・用量
転移性デスモイド抵抗性前立腺癌(mCRPC)
新規に診断された高リスクの転移性治療感受性前立腺癌(mHSPC)(内分泌療法を受けていない.または内分泌療法を受けてから最長3ヶ月経過している者を含む)。
5. [仕様]250mg
6.用法・用量
.
6.1.推奨用量
.
本剤の投与量は.1000mg(250mg×4錠)を1日1回経口投与することを推奨します。
本剤は.転移性除細動抵抗性前立腺癌(mCRPC)患者の治療において.プレドニゾンまたはプレドニゾロン5mgと併用して1日2回経口投与する。
本剤は.新規に診断された高リスクの転移性治療感受性前立腺癌(mHSPC)の治療において.プレドニゾンまたはプレドニゾロン5mgと併用し.1日1回経口投与する。
本剤の投与に際しては.ゴナドトロピン放出ホルモンアナログ(GnRHa)を併用していること.または両側睾丸摘出術を受けていることが必要です。 本剤は食前1時間以上.食後2時間以上の空腹時に服用すること(【薬物動態】の項参照)。 本製品は.水で丸呑みしてください。 分割したり.噛んだりしないでください。
投与中の毒性モニタリング
血清トランスアミナーゼは.本剤の投与開始前.投与開始後3カ月間は2週間ごと.その後は1カ月ごとに検査すること。 血圧.血清カリウム.体液貯留を毎月モニターする必要があります。 ただし.うっ血性心不全のリスクが高い患者では.投与開始後3ヶ月間は2週間に1回.その後は1ヶ月に1回の頻度でモニタリングを実施すること。
本剤投与前又は投与中に低カリウム血症を発症した患者では.患者の血中カリウム濃度を 4.0mM 以 下に維持するように注意すること。
患者が高血圧.低カリウム血症.浮腫または他の非サリコルチコイド毒性事象を含むグレード3以上の毒性事象を経験した場合.治療を中止し.適切な医学的管理を行う必要があります。 本剤の投与は.毒性の症状がグレード1またはベースラインレベルまで消失するまで.再開してはならない。
本剤.プレドニゾン又はプレドニゾロンの飲み忘れがあった場合には.翌日から通常の用量で治療を再開してください。
6.2.肝障害・肝毒性における用量調節の原則
肝機能障害。
ベースラインで軽度の肝障害がある患者さんでは.用量調節は必要ありません。
ベースラインで中等度の肝障害(Child-PughクラスB)がある患者には.本剤の推奨用量を1日1回250mgに減量すること。 ベースラインで中等度の肝障害(Child-PughクラスB)を有する患者における薬物動態試験では.本剤1000mgの単回経口投与により.アビラテロンの全身曝露量が約4倍増加した([薬物動態]を参照)。
中等度の肝障害のある患者では.アラニンアミノトランスフェラーゼ(ALT).アスパラギン酸アミノトランスフェラーゼ(AST)およびビリルビンの値を投与開始前.最初の1カ月は毎週.次の2カ月は2週間ごと.その後は1カ月ごとに観察する必要があります。 ベースラインで中等度の肝障害を有する患者において.ALT及び/又はASTが>5×正常上限(ULN).総ビリルビンが>3×ULN上昇した場合は.本剤を中止して再使用しないでください(【薬物動態】をご参照ください)。
本製品は.重度の肝障害(Child-Pugh クラスC)のある患者には使用しないでください。 また.別の試験では.ベースライン時に重度の肝障害(Child-PughクラスC)を有する8名の被験者と.肝機能が正常な8名の健常対照者を対象に.アビラテロンの薬物動態を分析しました。 ベースライン時に重度の肝障害があった被験者では.肝機能が正常な被験者と比較して.アビラテロンの全身曝露量(AUC)が7倍.遊離薬物画分の曝露量が2倍増加しました。
6.3.肝毒性
.
本剤投与中に肝障害(ALT及び/又はAST>5×ULN又は総ビリルビン>3×ULN)が発現した場合には.本剤の投与を一時的に中断し.用量を調節すること(【注意事項】を参照のこと)。 肝機能値がベースライン値に戻った後.またはASTおよびALT≦2.5×ULN.総ビリルビン≦1.5×ULNになった後.再投与のために750mg1日1回に減量することができる。 治療を再開する患者には.血清トランスアミナーゼ値及びビリルビン値を少なくとも2週間毎に.3ヶ月以降は毎月測定すること。
750mg1日1回投与で再び肝障害が発現した場合は.肝機能検査値がベースライン値に戻るか.ASTおよびALTが2.5×ULN以下.総ビリルビンが1.5×ULN以下になってから500mg1日1回に減量することができる。
1日1回500mgで再び肝障害が発生した場合は.本剤を中止する。
胆管閉塞やその他の原因でALTと総ビリルビンが同時に上昇していない場合.患者がALT>3×ULNを示し.総ビリルビン>2×ULNを伴う場合.本剤の投与を永久に中止すること。
6.4.腎機能障害時の投与量調整
について
腎障害のある患者には用量調節の必要はない([薬物動態]の項参照)。 しかしながら.重度の腎障害を有する前立腺癌患者における臨床経験はなく.そのような患者には注意が必要である。
6.5.強力なCYP3A4誘導剤との併用時の用量調整
本剤投与中は.強力なCYP3A4誘導剤(フェニトインナトリウム.カルバマゼピン.リファンピシン.リファブチン.リファペンチン.フェノバルビタール等)の併用は避けてください。 強力なCYP3A4誘導剤との併用が必要な場合は.併用期間中は投与回数を1日2回に増やす(例:1000mgを1日1回から1000mgを1日2回)。 併用する強力なCYP3A4誘導剤の投与中止後は.本剤を元の用量及び投与回数に調整すること([薬物相互作用]の項参照)。
高齢者
老人用】をご覧ください。
子供または青年
小児および青年に対する本製品の安全性および有効性は確立していない。
7.副反応
について
安全性プロファイルの概要
本剤は作用機序に起因する薬物動態作用により.高血圧.低カリウム血症.体液貯留を起こす可能性があります。 主な臨床的副作用は.末梢性浮腫.低カリウム血症.高血圧および尿路感染症です。 その他の重要な副作用には.心疾患.肝毒性.骨折.アレルギー性肺炎などがあります。 通常.塩コルチコステロイドの副作用は.効果的に管理することができます。 副腎皮質ステロイドの併用は.これらの薬物有害反応の発生率と重篤度を低減することができます。
7.1.臨床試験
7.1.臨床試験
7.
異なる薬剤の臨床試験で観察された副作用の発現率は.臨床試験の条件が異なるため.直接比較することはできず.臨床現場で観察される副作用の発現率を反映したものではありません。
mCRPC患者を対象とした2つの無作為化プラセボ対照多施設共同試験(COU-AA-301試験およびCOU-AA-302試験)において.治療群には本剤1000mgを1日1回.プレドニゾン5mgを1日2回併用投与されました。 対照群にはプラセボとプレドニゾン5mgを1日2回併用した。 3つ目の無作為化プラセボ対照多施設共同臨床試験(212082PCR 3011試験)は.高リスクのmHSPC患者を登録しました。 治療群には.本品1日1000mgとプレドニゾン(1日1回5mg)を併用した。 対照群の患者さんには.プラセボを投与しました。 また.mCRPC患者を対象とした2本の無作為化プラセボ対照試験(ABI-PRO-3001試験およびABI-PRO-3002試験)が実施されました。5本の無作為化対照試験で得られた2230例の安全性データの合計が.注意.グレード1~4の副作用およびグレード1~4の臨床検査の異常のデータの基礎となっています。 すべての試験において.両群ともGnRHaの投与または睾丸摘出術の既往があることが条件とされた。
プールデータでは.治療期間の中央値は.アビラテロン投与群で11ヵ月(0.1.43).プラセボ投与群で7.2ヵ月(0.1.43)でした。 臨床試験で報告され.アビラテロン投与群でより多く見られた(2%以上)副作用は.疲労.関節痛.高血圧.悪心.浮腫.低カリウム血症.ホットフラッシュ.下痢.嘔吐.上気道感染.咳および頭痛でした。
アビラテロン投与時の臨床試験で報告された臨床検査値異常は.貧血.アルカリホスファターゼ上昇.高トリグリセリド血症.リンパ球減少.高コレステロール血症.高血糖および低カリウム血症が最も多く(>20%).最も多い(≥2%)ものでした。
グレード3-4の有害事象は.アビラテロン投与群の53%.プラセボ投与群の46%で報告されています。 また.アビラテロン投与群の14%.プラセボ投与群の13%で投与中止が報告されています。 本剤およびプレドニゾンの投与中止に至った主な有害事象(1%以上)は.肝障害および心疾患であった。
治療中の有害事象に関連した死亡は.アビラテロン群で7.5%.プラセボ群で6.6%と報告されています。 アビラテロン群では.最も多い死因は病勢進行(3.3%)でした。 その他.肺炎.心停止.死亡(その他の情報なし).全身状態の悪化が5名から報告されている。
7.2 COU-A-301試験:ドセタキセル化学療法による前治療歴のある転移性デスモイド抵抗性前立腺癌
。
COU-AA-301試験は.ドセタキセルによる化学療法を受けたことのある転移性脱腫脹性前立腺癌患者1195名を対象に実施されました。 本試験では.肝転移のない患者については.ASTおよび/またはALTが2.5×ULN以上の場合は登録できないと規定した。 また.肝転移のある患者は.ASTおよび/またはALT>5×ULNの場合.登録に不適格とした。 表1は.COU-AA-301試験において.ベナドリル投与群でプラセボ投与群に比べて2%以上増加した有害事象及び特に懸念される有害事象の発現率を示したものである。 本剤とプレドニゾンの併用による治療期間の中央値は8ヵ月であった。
表1:301試験において.プラセボ群に比べアビラテロン酢酸塩群で2%以上高い確率で発現した有害反応または特別な注意を要する有害事象
| 本製品+プレドニン」と表記しています。 (N=791) |
Placebo + プレドニゾン Placebo + Prednisone (N=394) |
|||
| 全レベル1。 % |
3~4年生 % |
すべてのレベル % |
3~4年生 % |
|
| 筋骨格系および結合組織障害 | 30 26.2 |
4.2 3.0 |
23 23 |
4.1 2.3 |
| 全身性疾患 | 27 |
1.9 |
18 |
0.8 |
| 血管・リンパ管疾患 | 19 8.5 |
0.3 1.3 |
17 6.9 |
0.3 0.3 |
| 消化器系疾患 | 18 6.1 |
0.6 0 |
14 3.3 |
1.3 0 |
| 感染症・感染制御. 尿路感染症 上気道感染症 |
12 5.4 |
2.1 0 |
7.1 2.5 |
0.5 0 |
| 呼吸器.胸部および縦隔疾患。 咳 |
11 |
0 |
7.6 |
0 |
| 腎臓および尿路系疾患. 頻尿 夜間頻尿 |
7.2 6.2 |
0.3 0 |
5.1 4.1 |
0.3 0 |
| あらゆる種類の傷害.中毒および外科的合併症。 骨折5 |
5.9 |
1.4 |
2.3 |
0 |
| 心臓疾患 | 7.2 3.8 2.3 |
1.1 0.5 1.9 |
4.6 2.8 1.0 |
1.0 0 0.3 |
| 1 有害事象は.米国国立がん研究所の有害事象に関する共通用語基準(NCI CTCAE)バージョン3.0に従って等級付けされています。 2関節炎.関節痛.関節の腫れ.関節のこわばりなどの用語が含まれます。 3 :筋痙攣.骨格筋痛.筋痛.筋骨格系不快感.骨格筋緊張という用語を含む。 4 水腫.末梢水腫.脳震盪水腫.全身水腫という用語が含まれます。 5 病的骨折を除くすべての骨折を含む。 6 不整脈.頻脈.心房細動.上室性頻拍.心房頻拍.心室性頻拍.心房粗動.徐脈.完全房室ブロック.伝導障害および徐脈性不整脈という用語を含んでいます。 7狭心症.胸痛.不安定狭心症という用語が含まれています。 心筋梗塞または虚血は.本剤投与群よりもプラセボ群で多く報告されました(それぞれ1.3%.1.1%)。 <心不全.うっ血性心不全.左室機能不全.心原性ショック.心肥大.心筋症.駆出率低下などの用語が含まれます。 |
||||
表2は.COU-AA-301試験で懸念された臨床検査値異常を示したものである。
表2 COU-AA-301試験で懸念される臨床検査値異常について
| 本製品 + プレドニゾン (N = 791) | Placebo + Prednisone (N = 394) | |||
| 全レベル | 3年生~4年生 | 全レベル% | 3年生~4年生 | |
| 高トリグリセリド血症 | 63 | 0.4 | 53 | 0 |
| ASTの上昇 | 31 | 2.1 | 36 | 1.5 |
| 低カリウム血症 | 28 | 5.3 | 20 | 1.0 |
| 低リン酸血症 | 24 | 7.2 | 16 | 5.8 |
| ALT高値 | 11 | 1.4 | 10 | 0.8 |
| 総ビリルビン値上昇 | 6.6 | 0.1 | 4.6 | 0 |
。
7.3. COU-AA-302試験:化学療法未実施の転移性デバルキン抵抗性前立腺癌
7.4.
COU-AA-302試験は.前治療歴のない転移性腫瘍抵抗性前立腺癌患者さん1088名を募集しました。 肝転移を併発している患者は試験から除外され.ASTおよび/またはALTが2.5×ULN以上の患者も登録の対象外とされました。 表3は.COU-AA-302試験の治療群で5%以上に発現し.プラセボ群と比較して2%以上発現率が増加した有害事象を示したものである。 本剤とプレドニゾンの併用療法による治療期間の中央値は13.8カ月でした。
表3:COU-AA-302試験において.アビラテロン酢酸塩群の5%以上に発現し.プラセボ群より2%以上高い頻度で発現した有害事象
| 本製品 + プレドニゾン
. |
Placebo + プレドニゾン Placebo + Prednisone (N=540) |
|||
| 全レベル1。 % |
3~4年生 % |
すべてのレベル % |
3~4年生 % |
|
| 全身性疾患 | 39 25 8.7 |
2.2 0.4 0.6 |
34 21 5.9 |
1.7 1.1 0.2 |
| 筋骨格系および結合組織障害 | 30 6.6 |
2.0 0.4 |
25 4.1 |
2.0 0.7 |
| 消化器系疾患 | 23 22 11 |
0.4 0.9 0.0 |
19 18 5.0 |
0.6 0.9 0.2 |
| 血管・リンパ管疾患. ホットフラッシュ 高血圧症 |
22 22 |
0.2 3.9 |
18 13 |
0.0 3.0 |
| 呼吸器.胸部および縦隔疾患。 咳 呼吸困難 |
17 12 |
0.0 2.4 |
14 9.6 |
0.2 0.9 |
| 精神疾患. 不眠症 |
14 |
0.2 |
11 |
0.0 |
| あらゆる種類の怪我.中毒.手術の合併症。 打撲傷 フォールズ |
13 5.9 |
0.0 0.0 |
9.1 3.3 |
0.0 0.0 |
| 感染症・感染制御. 上気道感染症 上咽頭炎 |
13 11 |
0.0 0.0 |
8.0 8.1 |
0.0 0.0 |
| 腎臓および尿路系疾患. 血尿 |
10.3 |
1.3 |
5.6 |
0.6 |
| 皮膚および皮下組織の疾患… 発疹 |
8.1 |
0.0 |
3.7 |
0.0 |
| 1 有害事象はNCI CTCAEバージョン3.0に従って等級付けされています。 2末梢性浮腫.脳震盪性浮腫.全身性浮腫という用語が含まれます。 3関節炎.関節痛.関節の腫れ.関節のこわばりなどの用語が含まれます。 |
||||
表4は.COU-AA-302試験において15%以上の割合で発生し.プラセボ群に比べベナドリル投与群で高率(>5%)に発生した臨床検査値異常を示したものである。
表4:COU-AA-302試験において.アビラテロン酢酸塩群で15%.プラセボ群より高い確率(5%)で発生した臨床検査値の異常値
| 本製品+プレドニゾン(N=542) | プラセボ+プレドニゾン(N=540) | |||
| すべてのレベル % |
グレード 3-4 % |
すべてのレベル % |
3年生から4年生 % |
|
| 血液学. リンパ球減少症 |
38 |
8.7 |
32 |
7.4 |
| 血液生化学 | 57 42 37 33 17 |
6.5 6.1 3.1 0.4 2.8 |
51 29 29 25 10 |
5.2 0.7 1.1 0.2 1.7 |
| 1 非絶食時血液検査に基づく | ||||
。
7.4. 212082PCR 3011試験:mHSPCの高リスクの患者さんの治療
7.4. 212082PCR 3011試験:mHSPCの高リスクの患者さんの治療。
212082PCR 3011試験では.細胞毒性化学療法を受けたことのない.新たに高リスクのmHSPCと診断された患者さん1199名を募集しました。 肝転移を併発している患者は除外され.ASTおよび/またはALTがULNの2.5倍以上の患者も登録の対象外とされた。 すべての患者は試験中にGnRHaを投与されたか.または過去に両側睾丸摘出術を受けたことがある。 本剤とプレドニゾンの併用療法による治療期間の中央値は24ヶ月でした。
表5は.ベナドリル+プレドニゾン併用群の5%以上の患者に発現し.プラセボ+プレドニゾン併用群の発現率と比較して2%以上増加した有害事象を示したものである。
| Table5: Study:Table5: Table : 212082PCR 3011酢酸アビラテロン群における副作用で.プラセボ群に比べて≧5%.≧2%高い発生率。 strong>1 | ||||||||
| 本剤はプレドニゾン(N=597)と併用されている | プラシーボ(N=602) |
|||||||
| System/Organ Classification | 全レベル2 | 小学3年生~4年生 | すべてのレベル | 小学3年生~4年生 | ||||
| 副反応 | % | % | % | % | ||||
| 血管・リンパ管系疾患 | ||||||||
| 高血圧 | 37 | 20 | 13 | 10 | ||||
| ホットフラッシュ | 15 | 0.0 | 13 | 0.2 | ||||
| 代謝障害および栄養障害 | ||||||||
| 低カリウム血症 | 20 | 10 | 3.7 | 1.3 | ||||
| あらゆる種類の検査 | ||||||||
| ALT標高3 | 16 | 5.5 | 13 | 1.3 | ||||
| 東経高度3 | 15 | 4.4 | 11 | 1.5倍 | ||||
| 感染症・伝染病 | ||||||||
| 尿路感染症 | 7.0 | 1.0 | 3.7 | 0.8 | ||||
| 上気道感染症 | 6.7 | 0.2 | 4.7 | 0.2 | ||||
| あらゆる種類の神経障害 | ||||||||
| 頭痛 | 7.5 | 0.3 | 5.0 | 0.2 | ||||
| 呼吸器.胸部および縦隔疾患 | ||||||||
| cough4 | 6.5インチ | 0.0 | 3.2 | 0 | ||||
| 1 全例がGnRHa投与または睾丸摘出術を受けた。 2 有害事象はCTCAEバージョン4.0に従って等級付けされました。 3 有害事象または有害反応として報告されている。 4 咳嗽.咳痰.上気道咳嗽症候群を含む。 注:表6に記載された臨床検査値異常は.検査報告の値によって定義される。臨床検査値異常は.治験責任医師の見解により.臨床的に重要で.表5に記載された併用薬の投与または試験薬の調整を必要とする場合.有害事象として報告される。 |
||||||||
。
表6は.212082PCR3011試験において.15%以上の割合で発生し.ベンゼドリンとプレドニゾンの併用投与群でプラセボ群より高い割合(>5%)で発生した臨床検査値異常事象を示したものである。
。
7.5. 表6:本製品治療群の報告書フォームにおける臨床検査値異常>212082PCR 3011試験における15%の患者。 |
||||
| 本剤とプレドニンの併用について (N=597) |
プラシーボ | |||
| 臨床検査値異常 | 小学校1~4年生 % |
グレード 3-4 % |
1~4年生 % |
グレード 3-4 % |
| 血球数 | ||||
| リンパ球減少症 |
20 | 4 | 14 | 1.8 |
| 臨床生物化学 | ||||
| 低カリウム血症 | 30 | 9.6 | 6.7 | 1.3 |
| ALT標高 | 46 | 6.4 | 45 | 1.3 |
| 総ビリルビン値上昇 | 16 | 0.2 | 6.2 | 0.2 |
。
7.6. 注:表6に記載の臨床検査値異常は.検査報告の値によって定義されます。有害事象は.表5に記載の臨床検査値異常が臨床的に重要であると治験責任者が判断し.併用薬の投与または試験薬の調整を必要とした場合に報告されます。
.
7.7.重大な副作用の説明:
.
心血管系の有害事象
第3相試験(COU-AA-301試験およびABI-PRO-3001試験.COU-AA-302試験およびABI-PRO-3002試験.212082PCR 3011試験)では.コントロールされていない高血圧および臨床的に重大な心疾患(過去6ヶ月以内の心筋梗塞または動脈血栓塞.重症または不安定)を有する患者は除外されました。 狭心症.NYHA定義によるIII度またはIV度の心不全(COU-AA-301試験.ABI-PRO-3001試験).II度~IV度の心不全(212082PCR 3011試験.COU-AA-302試験.ABI-PRO-3002試験).心駆出力50%以上。 登録されたすべての患者さん(活性剤投与患者さん.プラセボ投与患者さんを含む)は.糖尿病.心筋梗塞.脳血管障害.心臓突然死と関連するGnRHaを主薬としたADTを併用されました。
5つの無作為化プラセボ対照臨床試験のプールデータでは.ベナドリル投与群の心不全発生率がプラセボ投与群より高かった(2.6% vs. 0.9%)。 グレード3-4の心不全がプラセボ群の1.3%に発生し.5名が治療を中断.4名が死亡した。 グレード3-4の心不全は.プラセボ投与群の0.2%に発生しました。 プラセボ群では心不全による死亡が2例発生し.治療中止の事象はなかった。
上記のプールデータでは.報告された不整脈の大半はグレード1~2であった。 プラセボ群では不整脈関連死が1例.突然死が3例.関連死が5例であった。 心停止による死亡は.投与群7例(0.3%).プラセボ群2例(0.1%)であった。 心筋虚血または心筋梗塞の発症による死亡は.プラセボ投与群で3例.プラセボ投与群で3例であった。
以下は.説明書の[注意事項]で詳しく説明しています。
- 塩分コルチコステロイドの過量投与による高血圧.低カリウム血症.体液貯留
- 副腎皮質機能不全
- 肝臓毒性
- 食品は本製品への曝露を増加させる可能性があります
。
市販後の経験
なお.本製品の販売承認後の使用において.自発的な報告に基づき.以下の追加副作用が確認されています。 頻度は.アンコモン≧1/1000および< 1/100, レア≧1/10,000および< 1/1000 である。
全身臓器分類:呼吸器.胸部.縦隔疾患
レア: アレルギー性肺胞炎
全身器官分類:様々な筋骨格系および結合組織系の障害
異常事態:横紋筋融解症.ミオパシー
全身臓器分類:肝胆膵系の疾患
稀なケース: 劇症肝炎.急性肝不全
副作用が疑われる場合の報告
医薬品が販売許可された後.副作用の疑いを報告することが重要です。 これにより.薬のベネフィットとリスクのバランスを継続的にモニターすることができます。 医療従事者は.副作用の疑いがある場合.国の副作用報告システムを通じて報告することが義務付けられています。
8.禁忌
8.禁忌
8.
- 本剤の有効成分または賦形剤に対して過敏症反応のある人は禁忌です。
- 妊娠中または妊娠の危険性のある女性には禁忌です。
- 重度の肝障害(Child-PughクラスC)のある患者には禁忌とされています。
。
。
9. [注意事項]
9.
9.1 塩分コルチコステロイド過剰投与による高血圧.低カリウム血症.体液貯留
.
本剤のCYP17阻害によるサルコルチコイドの濃度上昇により.高血圧.低カリウム血症.体液貯留が起こる可能性がある。 高血圧.低カリウム血症.体液貯留について.少なくとも月に1回.患者をモニターする。 本剤投与前および投与中は.高血圧をコントロールし.低カリウム血症を改善する必要があります。
プレドニゾン5mg1日2回とアビラテロン酢酸塩1000mg1日1回を併用した4本のプラセボ対照試験のプールデータによると.グレード3-4の低カリウム血症が治療群の4%.プラセボ群の2%の患者に認められました。 グレード3-4の高血圧は各治療群の2%に.グレード3-4の体液貯留は各治療群の1%に発現が認められました。
212082PCR 3011試験において.プレドニゾン5mg1日1回とアビラテロン酢酸塩1000mg1日1回の併用により.投与群の10%にグレード3-4の低カリウム血症.プラセボ群の1%にグレード3-4の高血圧が認められ.20%と10%にプラセボ群の高血圧が確認されました。 グレード3-4の体液貯留が全治療群の1%に発生しました。
心不全.最近の心筋梗塞.心血管疾患.心室性不整脈の患者など.基礎疾患の悪化につながる可能性のある血圧上昇.低カリウム.体液貯留のある患者には.本製品を使用して綿密なモニタリングを行う必要があります。 左室駆出率(LVEF) <50% またはNYHAクラスIIIまたはIVの心不全患者(COU-AA-301試験)またはNYHAクラスIIからIVの心不全患者(COU-AA-302試験および212082PCR3011試験)は臨床試験から除外されており.これらの患者に使用する本製品の安全性は不明である( 臨床試験]をご覧ください)。
9.2 副腎皮質機能不全
.
5つの無作為化プラセボ対照臨床試験のプールデータでは.本剤投与群2230名.プラセボ投与群1763名における副腎皮質機能不全の発生率はそれぞれ0.3%.0.1%であった。 本剤とプレドニゾンの併用療法を受けた患者において.毎日のステロイドの中止及び/又は感染症やストレスの併発により.副腎皮質機能不全が報告されています。 特に.プレドニゾンを中止した患者.プレドニゾンの投与量を減量した患者.異常なストレス状態を経験している患者において.副腎皮質機能不全の徴候や症状をモニターすること。 本治療による塩類コルチコステロイドの過量投与に伴う副作用は.副腎皮質機能不全の徴候や症状を覆い隠す可能性があります。 副腎皮質機能不全の診断を確定するために.臨床的に適切な検査を実施する。 副腎皮質ホルモンの投与量は.ストレス状況の発生前.発生中.発生後に増量する必要があるかもしれません。
9.3.肝毒性
9.3.肝毒性
9.3.1.
5つの無作為化臨床試験のプールデータによると.グレード3/4のALTまたはASTの上昇(少なくとも5×ULN)が本製品を投与された2230人の患者の6%に認められ.通常は投与開始後3ヶ月間に発生しました。 ベースラインのALTまたはASTが高い患者は.ベースラインの肝機能が正常な患者に比べ.肝機能マーカーが上昇する傾向があった。 本剤の治療群2230例のうち約1.1%がALT.ASTの上昇や肝機能の異常により投与を中止しています。 これらの臨床試験において.本製品に明らかに関連する肝毒性による死亡例は報告されていない。
治療開始前.治療開始後3ヶ月間は2週間ごと.その後は毎月.血清トランスアミナーゼ(ALT.AST)およびビリルビン値をモニターしてください。 ベースラインで中等度の肝障害を有する患者が低用量の250mgを投与される場合.投与開始前にALT.ASTおよびビリルビン値を監視し.投与開始1カ月間は週1回.次の2カ月間は2週に1回.その後は月1回監視すること。 肝毒性を示唆する臨床症状や徴候がある場合は.血清総ビリルビン値.ASTおよびALT値を速やかにモニターしてください。 AST.ALT.ビリルビンがベースラインより増加した場合.モニタリングの頻度を増やす必要があります。 ASTまたはALTが5×ULN以上.またはビリルビンが3×ULN以上上昇した場合には.本剤の投与を一時的に中止し.肝機能を注意深く観察する必要があります。
本剤の低用量での投与は.肝機能検査値が患者のベースライン値に戻った後.あるいはASTおよびALTが≦2.5×ULN.総ビリルビンが≦1.5×ULNになってから再開してください([用法]を参照)。 投与中のいずれかの時点で重篤な肝障害(AST または ALT≧20×ULN) が発現した場合には.本剤の投与を中止し.再投与を行わないこと。 販売後.まれに急性肝不全及び劇症肝炎(一部は致死的)が報告されている([有害事象]の項参照)。のように。
9.4.食品により本製品への曝露が増加する可能性がある
。
本製品は必ず空腹時に服用してください。 投与前2時間以上.投与後1時間以上絶食してください。 アビラテロンのCmax及びAUC0-∞(曝露量)は.空腹時の単回投与に比べ.食事と一緒に摂取することでそれぞれ最大17倍及び10倍増加します。 本剤と食品との反復併用投与による曝露量増加の安全性は評価されていない(【有害反応】及び【薬理作用】を参照)。
9.5.骨密度
.
進行性転移性前立腺癌(脱モアレスタンス性前立腺癌)の患者さんでは.骨塩量の減少が見られる場合があります。 この効果は.本製品とグルココルチコイドとの併用により増強される可能性があります。
9.6.ケトコナゾールの使用歴
ケトコナゾールによる治療歴のある前立腺癌患者は.寛解率が低くなる可能性があります。
9.7.高血糖
グルココルチコイドの使用は高血糖のリスクを高めるので.糖尿病の患者さんでは頻繁に血糖を測定する必要があります。
9.8.骨格筋の反応
本製品を使用した患者において.いくつかのミオパシー事象が報告されています。 一部の患者は腎不全を伴う横紋筋融解症を発症した。 ほとんどの症例は治療期間中の最初の1ヶ月間に発生し.製品中止後に回復しました。 本製品は.ミオパシー/横紋筋融解症に関連することが知られている薬剤を組み合わせて治療を受けている患者には.注意して使用する必要があります。
9.9. 化学療法併用療法
.
細胞障害性化学療法との併用における本剤の安全性及び有効性は確立していない。
9.10.賦形剤不耐性
本製品は乳糖を含んでいます。 ガラクトース不耐症.ラップ(Lapp)ラクターゼ欠損症.グルコース・ガラクトース吸収障害などのまれな遺伝的問題を持つ患者には投与しないこと。 また.本品には4錠あたり1.18mmol(または27mg)を超えるナトリウムが含まれています。 ナトリウムの摂取が制限されている患者さんでは.検討する必要があります。
9.11.その他の潜在的リスク
.
転移性デスモイド抵抗性前立腺癌の男性(本製品で治療中の方を含む)は.貧血や性機能障害のリスクがある可能性があります。
子供の手の届かないところに置いてください。
9.12. QT間隔
多施設共同オープン単群臨床試験において.mCRPC患者33名に本剤1000mgを1日1回食前1時間または食後2時間.プレドニゾン5mg1日2回と併用して投与しました。 ベースラインから2サイクル目の2日目までのQTc間隔に有意な変化は認められなかった(例:>20ms)。 ただし.臨床試験デザインの限界から.本製品がQTc間隔をわずかに延長する可能性を完全に否定することはできない(例えば.<10ms)。
機械の運転・操作能力への影響。
本製品は.運転や機械操作の能力に影響を与えないか.または無視できる程度である。
10.妊娠中・授乳中の方
10.1.妊活
本製品は.女性への使用は認められていません。 本剤の作用機序及び動物実験結果に基づき.胎児に障害を与え.妊娠を終了させる可能性があるため.妊娠中又は妊娠のおそれのある女性には禁忌とされています。
本製品の妊婦への使用に関するヒトでのデータはありません。 動物生殖試験において.器官形成期の妊娠ラットにアビラテロン酢酸塩を経口投与した場合.母体曝露量がヒト推奨用量における曝露量(AUC)の約0.03倍以上のとき.胎児発生に影響を及ぼすとされています。
10.2.母乳育児
10.2.
本製品は.女性への使用は認められていません。 本剤が母乳中に分泌されるかどうか.また.本剤が授乳期及び授乳中の乳児に及ぼす影響については不明である。
10.3.避妊
アビラテロンまたはその代謝物が精液中に存在するかどうかは不明です。 患者が妊婦と性行為をする場合は.コンドームが必要である。 妊娠可能な年齢の女性と性行為を行う場合は.コンドームの使用と他の有効な避妊方法が必要である。
動物生殖試験結果及び作用機序から.パートナーが妊娠可能な年齢の女性である男性には.本剤投与中及び最終投与後3週間は有効な避妊を行うよう指導する。
10.3.1.受胎可能期間
動物実験に基づき.本製品は生殖年齢にある男性の生殖能力を損なう可能性があります。
11. 【小児用】小児に対する有効性及び安全性は確立していない。
12.高齢者用
.
臨床試験で本製品が投与された患者のうち.70%が65歳以上.27%が75歳以上であった。 安全性と有効性については.高齢者と若年者の間で全体的な差は認められませんでした。 高齢者と若年者の本製品への反応性の違いを確認した臨床報告は他にないが.高齢者の感度が高いことを否定することはできない。
13. 【薬物相互作用】
.
他の薬剤との相互作用。
アビラテロンの曝露に対する他の薬剤の影響の可能性in vitro のデータから.本製品は CYP3A4 の基質であることがわかった。 健康人を対象とした薬物動態学的相互作用の臨床試験において.最初に強力なCYP3A4誘導剤であるリファンピシンを1日600mg.6日間投与し.その後本剤1,000mgを単回投与した場合.アビラテロンの平均血中AUC∞は55%減少しました。
強力なCYP3A4誘導剤(例:フェニトインナトリウム.カルバマゼピン.リファンピシン.リファブチン.リファペンチン.フェノバルビタール.セントジョーンズワート[Kanjiro])は.他に代替治療法がない限り治療中は回避すること。
健康人を対象とした薬物動態学的相互作用に関する別の臨床試験において.強力なCYP3A4阻害剤であるケトコナゾールとの併用は.アビラテロンの薬物動態に臨床的に意味のある影響を及ぼしませんでした。
アビラテロンの他の薬物への曝露に対する潜在的影響アビラテロンは.肝の薬物代謝酵素であるCYP2D6およびCYP2C8を阻害する。
本剤(+プレドニゾン)単回投与時のCYP2D6基質デキストロメトルファンへの影響を検討した試験において.デキストロメトルファンの全身曝露量(AUC)が約2.9倍増加しました。 デキストロメトルファンの活性代謝物である24のAUCは約33%増加した。
本剤とCYP2D6により活性化又は代謝される薬剤(特に治療域の狭い薬剤)との併用には注意が必要であり.治療域の狭い薬剤の減量を検討する必要がある。 CYP2D6で代謝される薬物には.メトプロロール.プロプラノロール.デシプラミン.ベンラファキシン.ハロペリドール.リスペリドン.プロパフェノン.フレカイニド.コデイン.オキシコドン.トラマドール(後の3剤はCYP2D6による活性鎮痛代謝物の生成を必要とします)などが含まれます。
健康人を対象としたCYP2C8による薬物相互作用試験において.本剤1,000mg単回投与でピオグリタゾンのAUCが46%増加し.ピオグリタゾンの活性代謝物M-Ⅲ及びM-ⅣのそれぞれのAUCが10%減少することが確認されています。 これらの結果から.主にCYP2C8によって排泄される薬剤と併用した場合.本剤の曝露量の増加は臨床的に重要ではないことが示唆されたが.2剤を併用する場合には.治療指標の狭いCYP2C8基質による毒性反応に注意することが必要である。
本剤の主要代謝物であるアビラテロン硫酸塩及びアビラテロンアジドは.トランスポータータンパク質OATP1B1の肝への取り込みを阻害するため.OATP1B1経由で排出される薬剤の濃度が増加する可能性がin vitro試験により示されています。 トランスポータータンパク質に基づく薬物相互作用に関する臨床試験のデータはありません。
QT間隔を延長することが知られている薬剤との併用。
デポ剤ではQT間隔が延長するため.クラスIA(キニジン.プロピザミド等)又はクラスIII(アミオダロン.ソタロール.ドフェチリド.イブリット等)の抗不整脈薬.メタドン.モキシフロキサシン.抗精神病薬等のQT間隔延長又は心室性先端捻転頻拍を誘発する薬剤と本品の併用は注意が必要である。
スピロノラクトンとの同時投与スピロノラクトンは.アンドロゲン受容体に結合し.前立腺特異抗原(PSA)値を上昇させる可能性があります。 本製品との併用は推奨しません。
14.オーバードーズ(Overdose)
。
本製品の過量摂取の経験は限られています。
本製品には特定の解毒剤はありません。 過量投与時には.本剤の投与を中止し.不整脈.心不全の監視.肝機能の評価等.総合的な支援策を講じること。
15. 【臨床試験】
.
本製品の有効性と安全性は.3つの無作為化プラセボ対照国際共同治験(COU-AA-301.COU-AA-302.212082PCR 3011試験)で証明されています。 これらの研究のすべての患者は.GnRHaを投与されているか.以前に両側睾丸摘出術を受けていた。 ケトコナゾールの投与歴のある患者.および副腎または下垂体疾患の既往のある患者は.これら3つの試験から除外された。 スピロノラクトンはアンドロゲン受容体と結合し.前立腺特異抗原(PSA)値を上昇させる可能性があるため.本製品の国際共同治験ではスピロノラクトンの使用は認められていません。
15.1.COU-A-301の学習
。
ドセタキセル化学療法による前治療歴のある転移性脱腫脹性前立腺癌患者
ドセタキセル化学療法による前治療歴のある難治性前立腺癌患者を対象とした無作為化プラセボ対照多施設共同第III相臨床試験において.本製品の有効性および安全性を評価すること。 合計1195名の患者を.1000mgを1日1回.プレドニゾン5mgを1日2回併用する経口投与(N = 797).またはプラセボを1日1回.プレドニゾン5mgを1日2回併用する経口投与(N = 398)に2:1の割合でランダムに割り付けました。 いずれかの群に無作為に割り付けられた患者は.病勢進行(プロトコルで定義された画像進行および症候性または臨床進行によるベースライン/最低値からのPSAの25%上昇と定義).新たな抗腫瘍療法の開始.忍容できない毒性または試験からの脱落まで治療を継続します。 前立腺癌に対して過去にケトコナゾールの投与を受けたことがあり.副腎または下垂体疾患の既往がある患者は.この試験から除外された。
患者背景とベースラインの疾患特性は.各群でバランスが取れていました。 登録患者の89%がEastern Collaborative Oncology Group(ECOG)フィジカルステータススコア0または1.45%がBrief Pain Scaleスコア4以上(過去24時間に報告された最も強い痛み)であった。 70%の患者は画像診断による病勢進行を認め.30%の患者はPSAのみによる病勢進行を認めた。 70%の患者は過去に1つの細胞毒性化学療法を受け.30%は両方のレジメンを受けていた。
552名の死亡が発生した後に予定されていたレジメン別の中間解析では.製品群の患者さんの全生存期間(OS)がプラセボ群に比べ統計的に有意に改善されました(表7.図1)。 775人の死亡が確認された後.生存率解析が更新された(最終解析では予定死亡数の97%)。 得られた結果は.中期分析結果(表7)と整合的であった。
表7:本剤またはプラセボとプレドニゾンまたはプレドニゾロンを投与した患者(GnRHaによる治療中または睾丸摘出術の既往)の全生存期間(intention-to-treat解析セット)。
| 。 生存データ解析 |
本製品+プレドニゾン本製品。 (N=797) |
Placebo+Prednisone (N=398) |
| 死者数 | 333(42%) | 219 (55%) |
| 全生存期間中央値(月) (95%信頼区間) |
14.8 (14.1, 15.4) | 10.9 (10.2, 12.0) |
| < 0.0001 | ||
| リスク比b(95%信頼区間) | 0.646 (0.543, 0.768) | |
| アップデートされた生存データ解析 | ||
| 死者数 | 501(63%) | 274 (69%) |
| 全生存期間中央値(月)(95%信頼区間) | 15.8 (14.8, 17.0) | 11.2 (10.4, 13.1) |
| 0.740 (0.638, 0.859) | ||
| a P値はlog-rank sum testに基づき.ECOGステータススコア(0または1)に応じて層別化されています。 b リスク比は層別化後の比例リスク比モデルによるものである。 リスク比率が<1であれば.優れた製品であることを示します。 |
||
。
治療開始後数カ月以内のすべての評価時点において.ベナドリル投与群の生存率はプラセボ投与群よりも高かった(図1)。
図1:本剤またはプラセボとプレドニゾンまたはプレドニゾロンを併用投与された患者(GnRHa治療中または睾丸摘出術前治療を併用)のカプラン・マイヤー生存曲線(Intention to treat解析セット)。
<テーブル
| -本製品 |
。
。
<テーブル
| プラシーボ」。 |
。
。
<テーブル
| 本製品 |
。
。
<テーブル
| 死亡時期(月) |
。
。
<テーブル
| 生存率(%) |
。
。
<テーブル
| 797 736 657 520 282 68 2 0 | 797 736 657 520 282 68 2 0 398 355 306 210 105 30 3 0。 ………. プラシーボ。 |
。
。
。
15.2.学習 COU-AA-302
.
化学療法を行わない転移性デバルキング抵抗性前立腺癌患者
本試験に登録された被験者は.化学療法を受けたことがなく.無症状または軽度の症状を有し.化学療法の臨床的適応をまだ有していない被験者でした。 Brief Pain Inventory(BPI-SF)によると.過去24時間の最も強い痛みの強さが0〜1点であれば無症状.2〜3点であれば軽症とされた。 中等度または中程度の痛みを持つ被験者.がん性疼痛にオピオイドを使用している被験者.内臓転移を持つ被験者は.本試験から除外されました。
合計1088名の患者を.ベナドリル1000mgを1日1回経口投与する群(n=546)とプラセボを1日1回経口投与する群(n=542)に1:1の割合でランダムに割り付け.両群にプレドニゾン5mgを1日2回併用投与しました。 患者は.画像上または臨床上(細胞毒性化学療法.放射線療法または外科治療.オピオイド治療またはECOGステータススコア3以上)の病勢進行.忍容できない毒性または試験からの脱落が生じた時点で治療を停止します。
本剤とプレドニゾンまたはプレドニゾロンの併用投与を受けた被験者の年齢の中央値は71歳.プラセボとプレドニゾンまたはプレドニゾロンの併用投与を受けた被験者の年齢の中央値は70歳であった。 民族別では.この治療群の被験者の520人(95.4%)が白人.15人(2.8%)が黒人.4人(0.7%)がアジア人.6人(1.1%)がその他であった。 両治療群において.76%の被験者がECOGフィジカルステータススコア0.24%がスコア1であり.50%の被験者が骨転移のみ.31%が骨転移と軟組織またはリンパ節転移.19%が軟組織またはリンパ節転移のみであった。 有効性の主要評価項目は.全生存期間と画像診断による無増悪生存期間(rPFS)としました。 これに加えて.オピオイドによるがん性疼痛の緩和までの時間.細胞毒性化学療法の開始までの時間.ECOGフィジカルステータススコアが悪化(ベースラインと比較して1点以上)するまでの時間.PSA増悪(前立腺がんワーキンググループ2[PCWG2]基準による)までの時間を評価指標として使用した。
画像診断による無増悪生存期間は.骨病変についてはPCWG2定義.軟部組織病変についてはmodified efficacy evaluation criteria for solid tumours( RECIST 1.1)などの連続画像診断を用いて評価した。rPFSは中央検査室でレビューした画像進行評価で解析された。
計画されたrPFS解析に基づき.画像上進行が確認された.あるいは死亡イベントが発生した被験者は合計401名で.ベンゼドリン投与群150名(28%).プラセボ投与群251名(46%)であった。 両治療群間でrPFSに有意差があった(表8.図2参照)。
表8:COU-AA-302試験:本剤またはプラセボとプレドニゾンまたはプレドニゾロンとGnRHaの併用投与または睾丸摘出術の既往がある患者における無増悪生存率
| 本製品+プレドニゾン」となります。 (N=546) |
Placebo+Prednisone. (N=542) |
|
| Progression-free imaging survival(rPFS) | ||
| 150 (28%) | 251(46%) | |
| rPFS中央値(月) | 満たしていない | 8.3 |
| (95%信頼区間) | (11.66;NE) | (8.12;8.54) |
| P値* | <0.0001 | |
| リスク比**(95%CI) | 0.425 (0.347; 0.522) | |
。
NE=未評価
*ベースラインのECOGフィットネスステータススコア(0または1)に対する層別log-rank検定によるp値
**リスク比率は1が有利です。
図2:本剤またはプラセボとプレドニゾン.プレドニゾロンとGnRHaの併用投与.睾丸摘出術前の患者における無増悪生存期間のカプラン・マイヤー曲線
<テーブル
。
。
OSの2回目の中間解析(IA)に先立ち.被験者データは引き続き収集された。 フォローアップ感度解析として治験責任医師が評価したrPFSの画像結果を表9と図3に示す。
画像進行または死亡は607名で.ベンゼドリン群271名(50%).プラセボ群336名(62%)であった。 ベナドリル投与群では.画像診断の進行または死亡のリスクがプラセボ投与群に比べて47%減少した(HR=0.530.95%CI: [0.451; 0.623].p<0.0001 )。 rPFSの中央値は.本製品群で16.5カ月.プラセボ群で8.3カ月でした。
表9:COU-AA-302試験:本剤またはプラセボとプレドニゾンまたはプレドニゾロンとGnRHaの併用投与または睾丸摘出術前の患者における無増悪生存期間(第2中間解析時・治験責任医師審査終了時)
| 本製品+プレドニゾン」となります。 (N=546) |
Placebo+Prednisone (N=542) |
|
| Progression-free imaging survival(rPFS) | ||
| 271(50%) | 336(62%) | |
| rPFS中央値(月) | 16.5 | 8.3 |
| (95%信頼区間) | (13.80;16.79) | (8.05;9.43) |
| P値* | <0.0001 | |
| リスク比**(95%CI) | 0.530 (0.451; 0.623) | |
。
*ベースラインのECOGフィットネスステータススコア(0または1)に対する層別log-rank検定によるp値
**危険率<1.そしてこのプロダクトはより有利である
図3:本剤またはプラセボとプレドニゾンまたはプレドニゾロンとGnRHaの併用投与または睾丸摘出術前の患者における無増悪生存期間のカプラン・マイヤー曲線(2回目のOS中間解析時・治験責任医師審査済)。
。 |
<テーブル
| 本製品 |
。
。
<テーブル
| 死亡時期(月) |
。
。
<テーブル
| 本製品 |
。
。
<テーブル
| プラシーボ |
。
。
333人の死亡が確認された後.予定されていたOS途中の解析が行われた。 本試験では.臨床的有用性が認められたため.非盲検化し.プラセボ群の被験者に本製品による薬物治療を提供しました。 プラセボ群に比べ.製品群では全生存期間が長く.死亡リスクも25%低かった(HR=0.752.95%CI: [0.606; 0.934].p=0.0097) が.OSの結果は未熟で.中間解析の結果は試験中止予定の統計的に有意なカットオフを満たさなかった(表10を参照)。 そのため.このIA後も被験者の生存を追跡調査した。
予定されていたOSの最終解析は.741名の死亡が確認された後に行われました(追跡期間中央値49ヶ月)。 ベナドリル投与群では65%(546例中354例).プラセボ投与群では71%(542例中387例)が死亡しました。 ベナドリル投与群では死亡リスクが19.4%減少し(HR=0.806; 95% CI: [0.697; 0.931], p=0.0033).OS延長効果は.OS中央値4.4カ月(ベナドリル投与群 34.7 ヶ月.プラセボ群 30.3 ヶ月;表10.図4参照)と統計的に有意であった。 プラセボ群では44%の被験者がその後本製品による治療を受けたが.それでも本製品群の臨床的有用性の優位性は顕著であった。
表10:COU-AA-302試験:本剤またはプラセボとプレドニゾンまたはプレドニゾロンとGnRHaの併用投与または睾丸摘出術の既往がある患者における全生存期間
| 本製品+プレドニゾン」。 (N=546) |
Placebo+Prednisone (N=542) |
|
| 生存期間に関する中間解析 | ||
| 147 (27%) | 186 (34%) | |
| 生存期間(中央値)(月) | 未達成 | 27.2 |
| (95%信頼区間) | (NE;ネ) | (25.95;NE) |
| P値* | 0.0097 | |
| リスク比**(95%CI) | 0.752 (0.606; 0.934) | |
| 生存率の最終解析 | ||
| 死亡率(%) | 354(65%) | 387 (71%) |
| 生存期間(中央値)(月) | 34.7 | 30.3 |
| (95%信頼区間) | (32.7;36.8) | (28.7;33.3) |
| P値* | 0.0033 | |
| リスク比**(95%CI) | 0.806 (0.697; 0.931) | |
。
NE=未評価
*ベースラインのECOGフィットネスステータススコア(0または1)に対する層別log-rank検定によるp値
** 本製品の優位性を示すリスク比率 <1
図4:本剤またはプラセボとプレドニゾンまたはプレドニゾロンとGnRHaの併用投与.睾丸摘出術の既往のある患者における生存率のカプラン・マイヤー曲線(最終解析)。
<テーブル
| 本製品 |
。
。
<テーブル
| プラシーボ |
。
。
<テーブル
| 本製品 |
。
。
<テーブル
| プラシーボ |
。
。
<テーブル
| 死亡時期(月) |
。
。
。
本製品は.全生存期間およびrPFSの改善に加えて.以下の副次的評価項目のすべてにおいて.プラセボ治療と比較して臨床的有用性を示しました。
PCWG2基準に基づくPSA増悪までの期間:PSA増悪までの期間中央値は.ベナドリル投与群で11.1ヵ月.プラセボ群で5.6ヵ月だった(HR=0.488.95% CI: [0.420; 0.568], p<0.0001)。 ベナドリル投与群では.プラセボ投与群に比べ.PSAの進行までの時間が約2倍長かった(HR=0.488)。 ベナドリル投与群では.プラセボ投与群よりも高い割合でPSAの寛解が証明された(62% vs 24%.p<0.0001)。 測定可能な軟部組織病変を有する被験者のうち.完全寛解または部分寛解に至った被験者の数は.ベナドリル投与群で有意に多く観察されました。
オピオイド使用までの期間:最終解析時のオピオイド使用までの期間中央値は.ベンゼドリン投与群33.4ヵ月.プラセボ群23.4ヵ月(HR=0.721.95% CI: [0.614; 0.846].p<0.0001 )でした。
細胞毒性化学療法開始までの期間:細胞毒性化学療法開始までの期間の中央値は.ベンゼドリン群で25.2カ月.プラセボ群で16.8カ月でした(HR=0.580.95% CI: [0.487; 0.691].p<0.0001)。
ECOG身体状態スコア≧1悪化までの期間:ECOG身体状態スコア≧1悪化までの期間は.ベナドリル投与群で12.3カ月.プラセボ群で10.9カ月でした(HR=0.821.95%CI: [0.714; 0.943].p=0.0053).
以下の試験結果は.本治療法の統計学的に有意な効果を反映しています。
客観的寛解率:RECIST基準(ベースラインのリンパ節サイズが2cm以上でないと標的病変とみなされない)に従って完全寛解または部分寛解を達成した測定可能病変を有する被験者の割合と定義した。 ベースラインで測定可能な病変を有する被験者のうち.客観的寛解を達成した割合は.治療群36%.プラセボ群16%であった(p<0.0001)。
痛み:ベナドリル投与群では.プラセボ投与群に比べ.痛みの強さの進行リスクの平均値が18%有意に減少しました(p=0.0490)。 痛みの強さが進行するまでの期間の中央値は.ベンゼドリン投与群で26.7ヶ月.プラセボ投与群で18.4ヶ月でした。
前立腺癌治療機能評価(FACT-P)(トータルスコア)進行までの時間:FACT-P(トータルスコア)の進行リスクは.プラセボ群に比べベナドリル投与群で22%減少しました(p=0.0028)。 FACT-P(総スコア)の悪化までの期間の中央値は.製品投与群で12.7カ月.プラセボ群で8.3カ月でした。
15.3. 212082PCR 3011試験(高リスクmHSPC患者に対する治療)
。
212082PCR 3011試験では.高リスクのmHSPC患者さん1199名を対象に.本剤1000mgをプレドニゾン(5mg/日)と併用で1日1回投与(N=597).またはプラセボ1日1回投与(N=602)を1対1の割合で無作為に割り付けました。 本試験に登録された患者さんは.無作為化前の治療を受けていない患者さんや.ADT(睾丸摘出術または抗アンドロゲン剤を含むGnRHa)による3ヶ月以内の治療を受けている患者さんを含む.新しくmHSPCと診断された患者さんです。 登録の1ヶ月前までに.緩和的放射線治療または緩和的手術(転移性疾患による症状の管理)を1コース受けることが許可された。 高危険度とは.ベースライン時に3つの危険因子のうち2つ以上(グリソンスコア8以上.骨スキャンで3つ以上の病変があること.測定可能な内臓転移があること)を有することと定義した。 重大な心機能障害.副腎機能障害.肝機能障害を有する患者は除外した。 患者さんは.画像または臨床的な疾患の進行.許容できない毒性.試験からの離脱.または死亡が起こるまで治療を続けます。 臨床的進行とは.がんに対する細胞毒性化学療法.放射線療法.手術が必要であること.オピオイドの長期使用を必要とする疼痛.ECOGフィジカルステータススコアが3以上に悪化したことと定義しました。
患者さんの属性は各群でバランスが取れていました。 年齢の中央値は67歳でした。 治療群の民族分布は.白人69%.黒人2.5%.アジア系21%.その他8.1%で.ECOGフィジカルステータススコアは.76%が0.42%が1.3.5%が2でした。 Brief Pain Questionnaire(過去24時間における最も強い痛み)の定義に基づき.ベースラインの痛みの評価が0〜1点(無症状)の患者が50%.2〜3点(軽症状)が23%.4点以上が28%。前立腺癌の治療経験がある患者は.手術(3.8%).放射線療法(3.8%).内分泌療法(93.2%)などであり.93.4%がこの分野の治療を受けていた . 内分泌療法は.GnRHaまたは拮抗薬(75.0%).睾丸摘出(12.0%).抗アンドロゲン療法(62.1%).エストロゲンおよびグルココルチコイド(1.4%)などであった。
有効性の主要評価項目は全生存期間です。 406名の死亡後.プロトコールの予定通り中間解析が行われ.ベナドリル+プレドニゾン併用療法群では.プラセボ群に比べ.統計的に有意にOSが改善したことが示された(表11.図5)。 ベナドリル+プレドニゾン併用療法群の21%.プラセボ群の41%が.細胞毒性化学療法.酢酸アビラテロン.エンザルタミド.全身放射線療法など.転移性CRPCのOS延長が期待できるフォローアップ治療を受けています。
| Table11: Study:11:11:11: 212082PCR 3011ベンゼドリンとプレドニゾンの併用投与群とプラセボ投与群における全生存期間(intention-to-treat解析セット) | ||
| 総合的な生存率 | 本製品とプレドニンの併用について (N=597) |
プラシーボ |
| 死亡 | 169(28.3%) | 237(39.4%) |
| 生存期間中央値(月) (95% CI) |
北緯60度 | 34.7(33.1.NE) |
| p値1 | <0.0001 | |
| リスク比2(95%CI) | 0.621 (0.509; 0.756) | |
| NE=Unable to Estimate(推定不能)。 1ログランク検定に基づくp値で.ECOGフィジカルステータススコア(0/1または2)と内臓転移の有無によって層別化した。 2リスク比は層別比例リスクモデルに基づいている。 リスク比が <1 の場合.プレドニゾンとの併用がより優れていることを示します。 |
||
。
図 5: Study 212082PCR 3011 Kaplan-Meier: 全生存期間 3011: 5: 5:全生存期間を延長する。 strong>plot; intention-to-treat population。
予定されていたrPFSの主要解析時点では.合計593件のイベントが発生し.ベンゼドリンとプレドニゾンの併用療法群では239人(40.0%).プラセボ群では354人(58.8%)が画像進行または死亡しました。 rPFSは.2つの治療群の間で有意差が認められました(表12.図6参照)。
| Table12:Study Table 12: Table 12: 212082PCR 3011ベンゼドリン併用プレドニゾン治療群とプラセボ治療群の画像無増悪生存率(Intention-to-treat Analysis Set) | ||
| Imaging Progression-Free Survival | 本製品とプレドニンの併用について (N=597) |
プラシーボ |
| 病気の進行または死亡 | 239 (40.0%) | 354(58.8%) |
| rPFS中央値(月) (95% CI) |
33.0 (29.57.NE) |
14.8 (14.69, 18.27) |
| <0.0001 | ||
| リスク比2(95%CI) | 0.466 (0.394; 0.550) | |
| NE=Unable to Estimate(推定不能)。 1ログランク検定に基づくP値で.ECOG体力スコア(0/1または2)により層別化した。 2リスク比は層別化後の比例リスクモデルに基づいている。 リスク比が <1 の場合.本製品とプレドニゾンの併用による ADT 治療が優れていることを示します。 |
||
。
全生存期間およびrPFSにおける有益性に加え.本製品とプレドニゾンの併用投与によるプラセボに対する有益性は.以下のように事前に定義されたすべての副次評価項目で観察されました。
<骨格関連イベントまでの期間: 骨格関連イベントのリスクを30%減少(HR = 0.703; 95% CI: [0.539, 0.916], p<0.0086). SREまでの期間の中央値は.今回のプレドニゾン併用療法群.プラセボ群ともにまだ到達していません。
PSA増悪までの期間(PCWG2基準による) Benzedrine併用療法群ではPSA増悪までの期間の中央値は.1.5年であった。 33.2ヵ月.プラセボ群7.4ヵ月(HR=0.299.95%CI: [0.255, 0.352].p<0.0001)。
追跡期間:中間解析の時点で.本剤とプレドニゾンの併用投与群では追跡期間の中央値に達していなかったのに対し.プラセボ群では21.6ヶ月(HR=0.415.95% CI: [0.346, 0.497], p<0.0001 )。
化学療法開始までの期間:化学療法開始までの期間の中央値は.本剤とプレドニゾンの併用投与群では未到達で.プラセボ群では38.9カ月(HR=0.443.95% CI: [0.349, 0.561].p<0.0001 )であった。
<痛みの進行までの期間:痛みの進行までの期間の中央値は.Benzedrineとプレドニゾンの併用療法群では到達しておらず.プラセボ群では16.6ヶ月でした(HR=0.695.95% CI: [0.583, 0.829], p<0.0001)。
ほとんどの探索的エンドポイントでは.本剤とプレドニゾンの併用療法がプラセボ療法より優れていることが示されました。
15.4. 中国人患者を対象とした臨床試験データ(ABI-PRO-3002試験)
15.5.
化学療法を行わない転移性デバルキング抵抗性前立腺癌患者
アジア(中国.マレーシア.タイ)および欧州(ロシア)の42試験施設において.無症状または軽症状の転移性デスモイド抵抗性前立腺癌患者を対象に.本剤とプレドニゾン/プレドニゾロン(以下.プレドニゾンと総称)の併用投与を無作為化二重盲検プラセボ対照第3相臨床試験で.被験者は地域(アジアまたは欧州)とECOGスコア(※)によって層別され.プレドニゾンと本剤の併用投与が行われました。 0または1)を層別化し.本剤とプレドニゾンの併用投与またはプラセボとプレドニゾンの併用投与のいずれかに無作為に割り付けた(1:1)。 対象者には.本剤1000mg(250mg×4錠)またはプラセボ4錠(1日1回)をプレドニゾン5mg(1日2回)と併用し.空腹時に投与しました。
本試験では.合計313名の被験者(157名:本剤とプレドニンの併用.156名:プラセボとプレドニンの併用)が登録され.そのうち238名の中国人患者が登録されました。 被験者は病勢が進行するまで治療を受けた。 本試験で定義された疾患進行とは.被験者がPSAの進行(直下点から25%以上増加.絶対増加量2ng/ml以上.3週間以上経過後に確認).画像進行(骨スキャン確認による進行.修正RECIST1.1基準による軟部組織疾患の進行).臨床進行(BPI-SF評価値4以上による進行)を経験したと治験責任者が判断した場合を含みます。 と骨格系の有害事象を伴う疼痛進行の確認.プレドニゾンの増量またはより強力なグルココルチコイドへの切り替え.または新しい全身性抗がん剤治療の開始)のいずれかを選択することができます。 また.被験者は.許容できない毒性または個人の選択により.治療を中止することができます。
有効性の評価には.血清PSA濃度による病勢進行の評価と生存率の評価が含まれます。 また.以下の評価も行った:全生存期間.客観的寛解率.併用薬または後続薬の記録.転移性前立腺がんに対する細胞毒性化学療法の開始までの期間.患者報告式質問票結果による疼痛進行までの期間.鎮痛剤使用スコアによる疼痛進行までの期間.病歴聴取および身体診察によるECOG身体状態における臨床悪化までの時間。
患者背景やベースラインの疾患特性は.両群間で概ねバランスがとれていました。 患者さんの年齢の中央値は71歳(48-90歳)でした。 プロトコルで予め定められた中間解析の時点までに.有効性の結果.本製品とプレドニゾンを併用投与した被験者では.プラセボとプレドニゾンを併用投与した被験者と比較してPSAの進行リスクが58%減少した(HR=0.418.p<0.0001)。中国人患者の結果は全体の結果と一致しており.本製品の投与により.PSA進行のリスクはプラセボとプレドニゾン併用投与群と比較して58%減少していることが示されました。 の被験者は.プラセボとプレドニゾンの併用療法群と比較して.PSAの進行リスクを44%減少させた(HR=0.563.p=0.0173)。
表13:ABI-PRO-3002試験のPSA増悪までの時間.層別解析(intention-to-treat解析セット)。
| —————– 中国 —————- | -総合 ————— | |||
| AA | プラシーボ | AA | プラシーボ | |
| 説明 a | (N=119) | (N=119) | (N=157) | (N=156) |
| ランダム化された被験者 | 119 | 119 | 157 | 156 |
| イベント | 30(25.2) | 43(36.1) | 34(21.7) | 60(38.5) |
| 切捨てられた尾部 | 89(74.8) | 76(63.9) | 123 (78.3) | 96(61.5) |
| p-valueb | 0.0173 | <0.0001 | ||
| リスク比(95%CI)c | 0.563(0.349;0.909) | 0.418 (0.271; 0.646) | ||
| a カプラン・マイヤー曲線に基づく評価。 b 地域(アジアまたはヨーロッパ)およびECOGスコアによる層別化したp値 c リスク比は層別比例リスク比モデルに基づいている。 リスク比率が <1 であれば.本製品は好ましい結果を示すと考えられます。 |
||||
。
図7:ABI-PRO-3002試験におけるPSA増悪までの時間のKaplan Meier曲線:全体(intention-to-treat解析セット)。
<テーブル
| この製品 プラセボ |
。
。
<テーブル
| この製品グループ —- プラセボ群 本品組合せ削除 ☆☆☆☆☆ プラセボ群削除 |
。
<テーブル
| ランダム時間(月単位) |
。
。
<テーブル
| 国名=全体 |
。
。
。
図8:ABI-PRO-3002試験におけるPSA増悪までの期間のKaplan Meier曲線:中国(Intention-to-treat解析セット)。
<テーブル
| この製品グループ —- プラセボ群 本品組合せ削除 ☆☆☆☆☆ プラセボ群削除 |
。
<テーブル
| 本製品はプラセボです |
。
。
<テーブル
| ランダム時間(月単位) |
。
。
<テーブル
| 国名=全体 |
。
。
。
PSAの寛解率は.本剤とプレドニゾンを併用した被験者(67%)が.プラセボとプレドニゾンを併用した被験者(31%)に比べ.有意に高かった(p<0.0001)。 中国人患者における結果は.全患者の結果と一致しており.PSA寛解率はベンゼドリンとプレドニゾンの併用投与群で67%.プラセボとプレドニゾンの併用投与群で37%であった(p<0.0001)。客観的寛解率(完全寛解および部分寛解.CR+PR)では.本剤とプレドニゾンを併用した被験者(23%)は.プラセボとプレドニゾンを併用した被験者(5%)に比べ.前者が約4.8倍と有意に高い寛解率が示されました(p=0.0369)。 寛解はすべて部分寛解であった。 中国人患者における結果は.全体集団における結果と一致しており.ベンゼドリンとプレドニゾンの併用治療群.プラセボとプレドニゾンの併用治療群における客観的寛解率はそれぞれ32%と0%でした(p=0.0052)。
安全性の結果.最も多く報告された有害事象(薬剤群およびプラセボ群の被験者の10%以上)は.骨痛.関節痛.背部痛.四肢痛および高血圧でした。 グレード3または4の有害事象は製品群で17%.プラセボ群で21%.重篤な有害事象は両群でそれぞれ4%.7%.死亡に至る有害事象はそれぞれ3%.4%.投与中止に至る有害事象はそれぞれ3%.5%が報告されました。
ABI-PRO-3002試験の結果.化学療法を受けていないmCRPC患者において.アビラテロン酢酸塩とプレドニゾンの併用療法が良好な臨床効果-リスク比を持つことが確認されました。 本試験における酢酸アビラテロン投与群の安全性プロファイルは.グローバルで実施された主要な第III相試験であるCOU-AA-302試験とほぼ一致しています。 新たな安全性シグナルは確認されませんでした。
15.5. 中国人患者を対象とした臨床試験データ(ABI-PRO-3001試験)
15.5.
ドセタキセル化学療法による前治療歴のある転移性減量抵抗性前立腺癌の患者さん
また.中国人患者を対象に.ドセタキセル化学療法が無効となった転移性減圧抵抗性前立腺癌患者を対象に.本剤とプレドニゾンを併用した第Ⅲ相ランダム化二重盲検プラセボ対照試験を実施しました。 すべての対象者は.1サイクル目の1日目に.アビラテロン酢酸塩1000mg(1日1回250mg×4錠)またはプラセボ(1日1回4錠)のいずれかと.プレドニゾン治療(1日2回5mg)を併用する群に2対1の比率で無作為に割り付けられます。 2つの治療群間のクロスオーバーは認められませんでした。 1回の治療サイクルは28日間でした。
本試験では.214名の被験者が登録され.無作為化されました。ITT解析および安全性解析には.酢酸アビラテロン群143名.プラセボ群71名が含まれました。 被験者は病勢進行が起こるまで治療された。 疾患の進行は.PSA の進行(Prostate Specific Antigen Working Group [PSAWG] 基準)および画像の進行(修正 RECIST 1.1 による骨スキャン進行または軟組織疾患の進行)と定義し.臨床進行(痛みの進行 [BPI-SF による評価].骨関連有害事象.プレドニンの増量.より強力なグルココルチコイドへの切り替え)の有無も問わないものとしました。 または.前立腺癌に関連する徴候・症状に対するオピオイド鎮痛剤の追加使用).または.PSAの増悪と画像の増悪の一方または両方を伴う臨床的な進行が認められる。
有効性は.試験期間中の被験者のPSA値を測定することにより評価した。 有効性の主要評価項目はTTPPで.無作為化からPSA増悪(PSAWG基準による)までの時間間隔と定義されました。 副次評価項目は.全生存期間.PSA寛解率.客観的寛解率.QOL総合スコアおよびFACT-Pサブスケールのスコア.疼痛進行までの時間.BPI-SF最悪疼痛強度スコアおよび疼痛薬使用スコアによる疼痛緩和が得られた被験者の割合.BFIによる疲労度の評価などです。
被験者の年齢の中央値は68歳であった。 被験者の大半は.ベースラインの病勢進行がPSAのみであった。 骨転移は.アビラテロン酢酸塩群で95.1%.プラセボ群で94.4%に認められ.ベースラインで疼痛症状があった被験者の割合は.それぞれ72.7%.66.2%でした。 すべての被験者が薬理学的または外科的なデバルキングを受けていた(61.7%が睾丸摘出術.54.7%がGnRHa治療を受けていた)。 すべての被験者が化学療法を受けていた。 二重盲検期間中の投与期間の中央値は.アビラテロン酢酸塩群で32.3週間.プラセボ群で16.9週間でした。 投与期間の中央値は.プラセボ群5サイクルに対し.アビラテロン酢酸塩群は9サイクルでした。 追跡期間中にアビラテロン酢酸塩のオープン投与を受けた被験者の治療期間の中央値は16.0週間.投与期間の中央値は4サイクル(16週間)であった。
有効性の結果.プラセボ群に比べ.アビラテロン酢酸塩群ではPSAの進行リスクが49%減少した(HR=0.506.p=0.0001)。 すべてのサブグループ解析において.ベースラインのECOGフィジカルステータススコアが2の被験者(サンプルサイズが小さいため)を除き.アビラテロン酢酸塩投与群ではTTPPが有意に改善されました。
。
図9:PSAWG基準に基づくランダム化からPSAまでの進行時間のカプラン・マイヤー・プロット図
(ABI-PRO- 3001試験:ITT解析セット)。
<テーブル
| プラシーボ |
。
。
<テーブル
| 本製品 |
。
。
<テーブル
| ランダム時間(月単位) |
。
。
<テーブル
。
。
。
ドセタキセルをベースとした化学療法が無効であった中国のmCRPC被験者において.酢酸アビラテロンとプレドニゾンの併用療法はTTPPを有意に改善し.高いPSA寛解率を達成しました。 また.OSについても臨床的に良好な傾向が認められ(HR=0.604 [0.356, 1.026] ).COU-AA-301試験と同様のHR(HR=0.646 [0.543, 0.768] )となりました。 PSAの寛解が確認された割合は.プラセボ群(14.1%.相対リスク3.525.p<0.0001)よりもアビラテロン酢酸塩群(49.7%)の方が有意に高いことが分かりました。 プラセボ群(50.7%)では.アビラテロン酢酸塩群(37.1%)と比較して.より多くの被験者が痛みの進行のイベントを経験しました。 アビラテロン酢酸エステルは.プラセボと比較して.痛みの進行リスクを50%有意に減少させました(HR=0.496; p=0.0014)。 疼痛スコアが4以上の被験者では.アビラテロン酢酸塩投与群の方が疼痛改善率が高く.両群の差は23%でした。
二重盲検期において.アビラテロン酢酸塩群ではグレード3~4の有害事象が32.2%.プラセボ群では28.2%.投与中に重篤な有害事象が14.0%と19.7%.投与中止に至る有害事象が7.0%と9.9%.投与中止に至る有害事象が6.3%と12.7%に報告され.プラセボ群と比べて.有害事象の発現率が高かった。 死亡に至る有害事象
ABI-PRO-3001試験の結果.化学療法を受けたMCRPC患者において.酢酸アビラテロンとプレドニゾンを併用した場合の良好なベネフィット・リスク比が確認されました。 本試験におけるアビラテロン酢酸塩群の安全性プロファイルは.グローバルでの主要な第III相試験(COU-AA-301試験)とほぼ一致していました。 新たな安全性シグナルは確認されなかった。
15.6. 212082PCR 3011試験における中国人被験者の臨床データ
15.6.
212082PCR 3011試験は.中国人被験者137名(本剤とプレドニゾン併用投与群:69名.プラセボ群:68名)が参加しました。 試験開始時.中国人被験者と全人口の人口統計および疾病特性は.治療群間で概ね均衡していた。 総治療期間の中央値は.Prednisol併用群で28ヵ月(31サイクル).プラセボ群で22ヵ月(24サイクル)であった。 サブグループ解析の結果は.サンプルサイズが小さいことから生じる偶然性の可能性があるため.慎重に検討する必要があることに留意することが重要である。
rPFSの主要解析時点において.ベナドリル+プレドニゾン併用療法群では18名(26.1%).プラセボ群では38名(55.9%)が画像進行または死亡を報告しています。 プラセボ群に比べ.プレドニゾン併用群では画像進行または死亡のリスクが66%減少した(HR = 0.341; 95% CI: 0.193, 0.605)。 ベンゼドリンとプレドニゾンの併用療法群ではrPFSの中央値は達成されず.プラセボ群ではrPFSの中央値は18.4カ月であった。
OSのハザード比は0.862(95%CI:0.415.1.788)であり.プラセボ群に比べ有意に高かった。 生存期間の中央値は.いずれの治療群でも達成されなかった。
中国人を対象とした有効性副次評価項目のサブグループ解析では.本剤とプレドニゾンの併用療法が優れている傾向が一貫して示されました。 プレドニゾンによる治療は.中国人被験者において化学療法開始の必要性を遅らせ(HR = 0.433.95% CI: 0.146, 1.279.化学療法開始までの時間中央値:いずれの治療群でも達成されなかった).追跡治療の必要性を遅らせ(HR = 0.349, 95% CI: 0.173, 0.707.追跡治療開始までの時間中央値:いずれの治療群でも達成されなかった).中国人被験者において追跡治療開始の必要性を遅らせ(HR = 0.349; 95% CI: 0.173, 0.707; 追跡治療開始までの時間中央値:いずれの治療群でも達成されなかった).追跡治療の開始を遅らせています。 中国人の痛みの進行遅延(HR = 0.680; 95% CI: 0.416, 1.111; 痛み進行までの期間中央値:プレドニゾン併用療法群では達成できず.プラセボ群では12.9ヶ月).中国人のPSA進行遅延(HR = 0.261; 95% CI: 0.157, 0.433; 進行までの時間中央値:プラセボ群:達成できず)。 (PSA悪化までの期間:ベンゼドリンとプレドニゾンの併用投与群では未達成.プラセボ群では9.2ヶ月)。
中国人の場合.ベンゼドリンとプレドニゾンの併用投与群では94.2%.プラセボ群では98.5%の被験者に治療上問題となる有害事象が報告されました。 最も頻繁に報告された有害事象(いずれかのグループの被験者の20%以上が報告)は.低カリウム血症(プレドニゾン併用群.プラセボ群でそれぞれ37.7%.20.6%).ALT上昇(30.4%.26.5%).AST上昇(27.5%.22.1%).高血糖(24.6%.20.6%).高血圧(24.6%.14.7%).背部高血圧症でした。 と14.7%).腰痛(10.1%と20.6%)であった。 グレード3または4の有害事象を報告した被験者の割合は両群でそれぞれ60.9%と50.0%.重篤な有害事象を報告した被験者の割合はそれぞれ23.2%と32.4%.投与中止に至った有害事象の報告者の割合はそれぞれ13.0%と16.2%.死亡に至った有害事象の報告者の割合はそれぞれ8.7%(すべて試験薬に関連していないと試験官により評価された)であります。 (いずれも治験責任医師が評価した試験薬との関連性はない).1.
212082PCR 3011試験で登録された中国人被験者に観察された本剤とプレドニゾン治療の併用による安全性プロファイルは.全体集団と概ね一致しており.新たな安全性の懸念は確認されませんでした。 212082PCR 3011試験では.中国で新たに高リスクのmHSPCと診断された患者さんの治療において.本剤とプレドニゾンの併用による良好なベネフィット・リスク比を示しました。
第16回 [薬理毒性学]
[薬物動態学
16.1.薬理作用
アビラテロン酢酸塩は.生体内でアビラテロンに変換され.精巣.副腎.前立腺腫瘍組織に発現し.アンドロゲン生合成に必要な17α-水酸化酵素/C17,20-リアーゼ(CYP17)を阻害するアンドロゲン生合成阻害剤である。
CYP17は.1)プレグネノロンとプロゲステロンを17α-水酸化酵素でそれぞれの17α-水酸化誘導体に変換し.2)その後C17,20リアーゼでそれぞれデヒドロエピアンドロステロンとアンドロステンジョンを生成する.2つの連続した反応を触媒している。 デヒドロエピアンドロステロンとアンドロステンジオンは.ともにアンドロゲンで.テストステロンの前駆体です。 また.アビラテロンによるCYP17の阻害は.副腎皮質塩基性ステロイドの産生を増加させることにつながる。
アンドロゲン感受性前立腺がんは.アンドロゲン低下療法に反応することがあります。 GnRHaや睾丸摘出などのアンドロゲン遮断療法は.精巣でのアンドロゲン産生を減らすことができますが.副腎や腫瘍でのアンドロゲン産生には影響を与えることができません。
プラセボ対照臨床試験において.酢酸アビラテロンは.患者さんの血清テストステロンおよびその他のアンドロゲン値を低下させました。 臨床使用において.本製品の血清テストステロン値への影響を監視する必要はない。
血清PSA値は変動する可能性があるが.個々の患者における臨床的有用性と相関することは示されていない。
16.2.毒性試験
反復投与毒性:ラットの13週及び26週.サルの13週及び39週の反復投与毒性試験において.アビラテロン酢酸塩はヒト臨床曝露量の約半分の用量(AUC)で循環テストステロン値の減少をもたらしました。 その結果.雄雌の生殖器.副腎.肝臓.下垂体(ラットのみ).雄乳腺において.臓器重量の減少や何らかの毒性が認められました。 生殖器官の変化は.アビラテロン酢酸塩の抗アンドロゲン薬理作用と一致するものであった。 ラットにおいて.アビラテロン酢酸塩を50 mg/kg/日以上(ヒトのAUCに近い)の用量で毎日経口投与したところ.用量依存的に白内障の発生率が増加しました。 酢酸アビラテロンの連日経口投与によるサル39週間試験では.高用量(ヒトAUCの2倍)でも白内障は観察されなかった。
遺伝毒性:アビラテロン酢酸及びアビラテロンエームス試験.ヒトリンパ球細胞遺伝試験.ラット小核試験の結果は全て陰性であった。
生殖毒性: 雄ラット(13週及び26週)及びサル(39週)に50mg/kg/日以上(ラット)及び250mg/kg/日以上(サル)の用量で反復投与した毒性試験で.生殖機能の萎縮.無精子症/精子減少及び増殖性変化が認められ.アビラテロンの抗アンドロゲン薬理作用と一致する作用が認められました。 これらの作用を示すAUCは.ラットではヒトの臨床曝露量付近.サルでは約0.6倍で観察された。
ラット生殖能及び初期胚発生毒性試験において.30mg/kg/日及びそれ以上の用量を4週間投与した雄ラットで.生殖器官重量の減少.精子数の減少.精子生存率の低下.精子形態の変化.生殖能力の低下が見られた。 未投与の雌ラットに30 mg/kg/日を投与した雄ラットを交配させたところ.黄体数の減少.出生・生存胚の減少.着床前の喪失率の上昇がみられた。 雄ラットの生殖能力に対するアビラテロン酢酸塩の影響は.16週間の休薬で回復した。 雌ラットに30 mg/kg/日以上のアビラテロン酢酸塩を交配2週間前から妊娠7日目まで投与したところ.発情周期の不順又は遅延の発生率及び前期死亡率の増加が認められた(300 mg/kg/日)。 アビラテロン酢酸塩を投与した雌ラットの交配能力.受胎能力およびその子孫のパラメータに差は認められなかった。 雌ラットのアビラテロン酢酸塩の作用は.4週間の投与中止で回復した。 ラットの30 mg/kg/日の用量は.体表面積換算でヒトの推奨用量(1000 mg/日)の約0.3倍となる。
ラット胚・胎児発生毒性試験において.妊娠6~17日目にアビラテロン酢酸塩を10.30及び100 mg/kg/日(それぞれヒトAUCの約0.03.0.1及び0.3倍)で経口投与すると発生毒性を示し.10 mg/kg/日以上で胚・胎児死亡(到着後損失及び胎児吸収率の増加.生存胎児数減少)が認められました。 30mg/kg/dose以上で胚発生遅延(骨格)及び両側尿管拡張.胎児の肛門性器距離短縮.100mg/kg/doseで胎児体重減少が認められた。 10mg/kg/回以上の投与では母体毒性を引き起こす可能性がある。
発がん性: ラットを用いた2年間の経口がん原性試験において.アビラテロン酢酸塩を雄に5.15.50mg/kg/日.雌に15.50.150mg/kg/日の用量で投与したところ.すべての用量で精巣間葉系細胞腺腫および間葉系細胞がんが認められ.アビラテロンの薬理活性と関連すると考えられています。 Abiraterone acetateは.ヒトの0.8倍の曝露量で雌マウスに発がん性が認められませんでした。 Tg.rasH2トランスジェニックマウスの6ヶ月間発がん性試験において.発がん性は認められませんでした。
第17回 [薬物動態]
について
本剤および活性代謝物であるアビラテロンの薬物動態は.健常人およびmCRPC患者を対象に検討されています。 生体内では.速やかにアビラテロンに変換される。 臨床試験では.99%の検体で本製品の血漿中濃度が検出レベル(0.2ng/ml)未満であった。
17.1.吸収量
.
mCRPC患者において.本製品を経口投与した後.アビラテロンのピークまでの時間の中央値は2時間でした。 アビラテロンの定常蓄積は.本剤1000mg単回投与時の2倍の曝露量(定常AUC)で確認された。
mCRPC患者において.1日1回1000 mg投与時のCmaxおよびAUC定常値(平均±SD)はそれぞれ226 ± 178 ng/mlおよび993 ± 639 ng/h/mlであり.250~1000 mgの用量範囲で用量比例からの大きな逸脱は認められなかった。 1000 mgから2000 mgに増量した場合,曝露量の有意な増加は認められなかった(平均AUCは8%増加)。
本製品を食品と一緒に投与した場合.アビラテロンの全身曝露量が増加した。 アビラテロンのCmax及びAUC0-∞は.本剤と低脂肪食(脂肪分7%.カロリー300kcal)の併用によりそれぞれ約7倍及び約5倍に増加し.本剤と高脂肪食(脂肪分57%.カロリー825kcal)の併用によりそれぞれ約17倍及び10倍に増加しました。 食品の多様性及び変動性を考慮すると.本製品と食品との併用により.曝露量が増加し.変動する可能性がある。 そのため.服用前2時間以上.服用後1時間以内は食べ物を摂取しないようにしてください。 また.本剤は全錠で水と一緒に服用すること(【用法・用量】を参照)。
17.2.分布と蛋白質結合
.
アビラテロンは.ヒト血漿タンパク質であるアルブミンおよびα-1酸性糖タンパク質と高い結合率(>99%)を有しています。 定常状態の見かけの分布容積(平均±SD)は 19,669 ± 13,358 Lであった。in vitro試験において.本剤及びアビラテロンは臨床的に適切な濃度範囲においてP糖蛋白質の基質ではなく.本剤はP糖蛋白質に対する阻害剤であることが示された。
17.3.メタボリズム
.
14C-アビラテロン酢酸塩カプセルを経口投与すると.アビラテロン酢酸塩はアビラテロン(活性代謝物)に加水分解される。 この過程は.おそらくエステラーゼの作用によって変換され(エステラーゼは同定されていない).CYPを介することはない。 ヒト血漿中のアビラテロンの2つの主要な循環代謝物は.硫酸アビラテロン(不活性)とN-酸化硫酸アビラテロン(不活性)で.それぞれが曝露量の約43%を占めています。CYP3A4とSULT2A1はN-酸化硫酸アビラテロンの生成に関与し.SULT2A1もまた硫酸アビラテロンの生成に関与しています。
17.4.排泄
17.4.排泄
17.
mCRPC患者におけるアビラテロンの血漿中終末半減期(平均±SD)は.12時間±5時間でした。 14C-アビラテロン酢酸塩の経口投与後.糞便および尿からそれぞれ約88%および5%の放射性物質が回収された。 糞便中に存在した主な化合物は.製品の原型およびアビラテロン(それぞれ投与量の55%および22%)であった。
17.5.肝障害のある患者
ベースライン時に軽度の肝障害(Child-PughクラスAおよびB)を有する被験者(n = 8)および肝機能が正常な健康な被験者(n = 8)において.アビラテロンの薬物動態が評価されました。 ベースライン時に軽度および中等度の肝障害を有する被験者において.1000mgを空腹時に単回経口投与した場合.アビラテロンの全身曝露量はそれぞれ約1.1倍および3.6倍に増加しました。 アビラテロンの平均半減期は.軽度および中等度の肝障害を有する被験者において.それぞれ18時間および19時間に延長されました。
また.別の試験では.ベースライン時に重度の肝障害(Child-PughクラスC)を有する8名の被験者と肝機能が正常な8名の健常者を対象に.アビラテロンの薬物動態を分析しました。 ベースライン時に重度の肝障害を有する被験者では.肝機能が正常な被験者と比較して.アビラテロンの全身曝露量(AUC)が約7倍増加しました。 また.本試験では.ベースライン時の平均タンパク質結合率が.肝機能が正常な被験者に比べて重度の肝障害を有する被験者では低く.その結果.遊離薬物画分への曝露量が重度の肝障害を有する被験者では2倍に増加することが判明しました。 (用法・用量]の項参照
17.6.腎臓障害のある患者
.
末期腎不全患者(n=8)および安定した血液透析を受けている腎機能正常者(n=8)を対象に.アビラテロンの薬物動態が評価されました。 末期腎不全患者群では.透析終了1時間後に本剤1000 mgを空腹時に単回経口投与し.投与後96時間以内にサンプリングして薬物動態解析を実施した結果.本剤の投与量は1,000 mg/日であった。 その結果.透析を受けている末期腎不全の被験者では.1000mgを単回経口投与しても.腎機能が正常な被験者と比較してアビラテロンの全身曝露量の増加は認められなかった(【注意事項】を参照)。
第18回 [ストレージ]
[ストレージ]15〜30℃の間で保管してください。
第19回 【パッケージ】高密度ポリエチレン丸型ボトル.120錠/ボトル
20. [有効期限]
.
24ヶ月
21. [エグゼクティブ・スタンダード]
21.
JX20130141
22.輸入医薬品登録証番号]
。
H20150264
第23回 [メーカー]
.
会社名:パセオン株式会社
生産拠点住所:カナダ オンタリオ州ミシサガ市 2100 Syntex Court, L5N 7K9, Canada
23.1.国内の連絡先情報
名称:西安ヤンセンファーマ株式会社(以下.「西安ヤンセンファーマ」という。
住所:西安市高新技術区曹洞科学技術産業基地曹洞四路19号
郵便番号:710304
電話番号:400 888 9988
ファックス番号:(029) 82576616
ウェブサイト:http://www.xian-janssen.com.cn