承認日
オキシチニブメシル酸塩錠の用法・用量。
使用上の注意をよく読み.医師の指導のもとに使用してください
。
[薬名]。
一般名:オシチニブメシル酸塩錠
販売名:TERRISSA®/TAGRISSO®。
英語名:Osimertinib Mesylate Tablets
羽生ピンイン: Jiahuangsuan Aoxitini Pian
【成分】
本製品の有効成分はオシメルチニブメシル酸塩である
化学名:N-{2-{[2-(ジメチルアミノ)エチル](メチル)アミノ}-4-メトキシ-5-{[4-(l-メチル-lH-インドール-3-イル)ピリミジン-2-イル]アミノ}フェニル)プロパン-2-エナミドメタンスルホネート
化学構造式。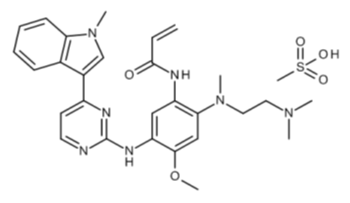
分子式:C28H33N7O2 ・・・・・・・・・・・・・。CH4O3S。
分子量:595.71
[Properties]。
本品は淡褐色のフィルムコーティング錠で.コーティングを剥がすと白色から淡褐色になります。
オシチニブメシル酸塩40mg:片面に「AZ」「40」の文字が印刷され.もう片面は白紙です。
オシチニブメシル酸塩 80mg:片面に「AZ」「80」の文字が印刷され.もう片面は白紙です。
[効能・効果]。
【効能・効果】
本剤は.上皮成長因子受容体(EGFR)チロシンキナーゼ阻害剤(TKI)による前治療で病勢が進行した.または治療後にEGFR T790M変異が陽性の局所進行性および転移性非小細胞肺癌(NSCLC)の成人患者の治療を適応としています。
【スペック】
【用法・用量】
本製品は.抗悪性腫瘍剤治療に経験豊富な医師が処方する必要があります。
局所進行性または転移性NSCLCに本製品を使用する前に.まずEGFR T790M変異の状態を明らかにする必要があります。 EGFR T790M変異の有無は.本剤投与前に十分に検証された検査で判断すること(詳細は[使用上の注意]を参照)。
用法・用量。
本剤の推奨用量は.疾患の進行または忍容性のない毒性が発現するまで.1日80mgとする。
本剤を1回でも服用しなかった場合.次の服用が12時間以内でなければ.本剤を補う必要があります。
本製品は.毎日同じ時間に.食事と一緒に.または空腹時に摂取してください。
投与量の調節。
なお.個々の患者の安全性及び忍容性に応じて.投与量を一時停止又は減量することができる。 減量が必要な場合は.1日1回40mgまで減量すること。
有害事象(AE)および毒性の発現に伴う減量の原則については.表1を参照してください。
表1.オシチニブメシル酸塩錠の有害事象発生後の投与量調整原則
| AdverseEventa | 投与量調節 | |
| Lungs | 間質性肺疾患/非感染性肺炎 | 本製品の販売を永久に中止すること |
| Heart | 少なくとも2回の心電図検査でQTc間隔が500ms以上であることが示唆されていること | QTc間隔が481ms未満になるまで.またはベースラインに戻るまで本剤の投与を一時中止すること(ベースライン値が481ms以上の場合.40mgの用量で投与を再開すること) | QTc間隔が481ms未満になるまで本剤の投与を一時中止すること。 |
| 重篤な不整脈の徴候または症状を伴うQTc間隔の延長 | 本製品の販売を永久に中止すること | |
| 無症状の絶対的左室駆出率(LVEF)がベースラインに対して10%以上50%未満低下しているもの | 最大4週間.本治療を中止する。 – ベースラインのLVEFレベルまで改善されれば.治療を再開する。 – ベースラインレベルまで改善が見られない場合.治療を永久に中止する。 |
|
| 症候性うっ血性心不全 | 本剤の投与を永久に中止すること。 | |
| Other | グレード3以上の副作用 | 最大で3週間の使用停止 |
| グレード3以上の副作用が.3週間以内の本製品の使用中止によりグレード0~2に改善された場合 | その後.元の用量(80mg)または減量した用量(40mg)で再開することができます。 | |
| 本剤を最長3週間休薬しても.グレード3以上の副作用がグレード0~2に減少しない場合 | 本製品の販売を永久に中止すること |
。
a 注:臨床有害事象の強度は.米国国立がん研究所(NCI)の有害事象共通用語基準(CTCAE)バージョン4.0に従って等級付けされています。
QTc:心拍数で補正したQT間隔
特別な人々。
患者の年齢.体重.性別.人種.喫煙状況による用量調節は必要ない(【薬物動態】の項参照)。
肝機能障害。
軽度の肝障害(総ビリルビン値および正常上限値.グルタミン酸トランスアミナーゼ(AST)値が1~1.5×ULN.または総ビリルビン値が1~1.5×ULN.AST値が無制限)患者においては用量調節は必要ないが.そのような患者においては慎重に使用すること。 中等度から重度の肝障害患者における本剤の安全性及び有効性は不明である。 中等度から重度の肝障害のある患者には.より多くの情報が得られるまで使用することは推奨されません。(薬物動態]参照)。
腎臓障害。
軽度から中等度の腎機能障害を有する患者への本製品の使用において.用量の調節は必要ない。 重篤な腎障害を有する患者における本製品の使用に関するデータは限られています。 末期腎不全患者(Cockcroft and Gault式<15mL/minで算出したクレアチニンクリアランス(CLcr))及び透析中の患者における本剤の安全性及び有効性は不明である。 本剤は.重度又は末期の腎障害のある患者には注意して使用すること([薬物動態]の項参照)。
投与方法。
本製品は経口用です。 本製品は.丸ごと水と一緒に服用し.粉砕.破損.噛み砕くことはできません。
患者が薬を飲み込めない場合は.錠剤を50mLの無炭酸水に溶かして使用することができる。 錠剤は砕かずに水に落とし.そのまま分散するまでかき混ぜ.素早く飲み込む。 さらにコップ半分の水を加えて飲み残しがないようにし.素早く飲み込む。 他の液体を加えてはいけません。
胃管で栄養補給する場合.15mLの水を薬剤の最初の溶解に使用し.その後の残留物の洗浄に15mLを使用する以外は.上記と同様に行うことができる。これらの30mLの液は.経鼻胃管の製造業者の指示に従って供給し.適量の水で洗浄すること。 これらの溶解液.残液ともに錠剤を投入後30分以内に投与する。
【副作用】。
安全性データの概要(因果関係を考慮しない)。
T790M変異陽性の前治療歴のあるNSCLC患者411名を対象に.1日80mgの用量を投与した2つの国際共同単群臨床試験(AURA拡張試験の第II相部分およびAURA2試験)において安全性データが得られており.411例中333例が6カ月以上.97例が9カ月以上本剤による曝露を受け.曝露例はゼロでした。 411人の患者のうち.333人は少なくとも6ヶ月間.97人は少なくとも9ヶ月間.しかし.12ヶ月間まで治療を受けている患者はいなかった。
投与群で最も多かった有害事象(20%)は.下痢(42%).発疹(41%).皮膚乾燥(31%)および手足の爪の毒性(25%)でした。
減量または投与中止に至った主な有害事象は.心電図QTc間隔の延長(2.2%)および好中球減少(1.9%)でした。2%以上の患者さんに報告された重篤な有害事象は肺炎と肺塞栓症でした。 その他.感染性肺炎(4名).心血管系事故・脳出血(2名)など.1名以上の患者において致命的な有害事象が報告されています。 治療群の5.6%で有害事象により治療が中断された。 投与中止に至った主な有害事象は.間質性肺疾患/非感染性肺炎.脳血管障害/脳梗塞であった。
表2 発生率>>10% 2つのグローバル単群試験におけるNCICTCAE*の発生状況
. strong>グレードの高い有害事象と発生率>2% NCI CTCAE* Grade 3-4 Adverse Events CTCAE* Grade 3-4
| AdverseEvent | オキシチニブ. N=411 |
|
| 全学年 | 3~4 f> | |
| % | % | |
| 消化器系疾患 | ||
| 下痢 | 42 | 1.0 |
| ナスティ | 17 | 0.5 |
| ナガス | 16 | 0.7 |
| 便秘 | 15インチ | 0.2 |
| 口内炎 | 12 | 0 |
| 皮膚病 | ||
| ラッシュa | 41 | 0.5 |
| 乾燥肌 b | 31 | 0 |
| 指(足指)爪の毒性 c | 25 | 0 |
| かゆみ | 14 | 0 |
| 眼病 d | 18 | 0.2 |
| 呼吸器系疾患 | ||
| 咳き込む | 14 | 0.2 |
| 全身性疾患 | ||
| 疲労 | 14 | 0.5 |
| 筋肉肉系疾患 | ||
| 背中の痛み | 13 | 0.7 |
| 中枢神経系 | ||
| 頭痛 | 10 | 0.2 |
| 感染症 | ||
| 4 | 2.2 | |
| 血管イベント | ||
| 7 | 2.4 | |
。
* NCI CTCAE v4.0。
- 発疹様分類用語として.発疹.全身発疹.紅斑性発疹.黄斑性発疹.丘疹性発疹.膿疱性発疹.紅斑.毛包炎.ざ瘡.皮膚炎.ざ瘡状皮膚炎が含まれる報告症例を対象としています。
- 乾燥肌.湿疹.ひび割れ.乾燥性疾患など。
- 以下の分類された用語の報告例を含む:爪床疾患.爪床の炎症.爪床の圧痛.爪床の変色.指(足)爪疾患.指(足)爪の毒性爪.指(足)爪の萎縮.指(足)爪の感染.指(足)爪の硬化.脆性爪.爪剥離.脱爪症.爪真菌。
- 目の乾燥.目のかすみ.角膜炎.白内障.目やに.眼瞼炎.目の痛み.涙の増加.飛蚊症などを含む。 その他の眼毒を呈している患者さん<1%。
- 深部静脈血栓症.内頸静脈血栓症.肺塞栓症などを含む。
- グレード4の事象は報告されていません。
。
。
。 安全性データの概要(明らかに副作用と思われる部分)。
本製品を服用した患者さんにおける一般的な副作用(ADR)の発現率を表3に示しました。
ADRは.MedDRAのSystemic Organ Classification(SOC)に従って集計されています。 各SOC内では.発生頻度の高いADRを筆頭に順位付けを行いました。 各頻度区分の中で.ADRは重症度の降順にランク付けされた。 また.各ADRの対応する頻度は.従来のCIOMS IIIの概念に従って分類され.これらの頻度区分は.非常に多い(≧1/10).多い(>1/100〜<1/10).少ない(≧1/1,000〜<1/100).少ない(≧1/10,000〜<1/1000)であった。 非常に稀 (<1/10,000); 不明 (利用可能なデータに基づいて推定できない)。 本項では.患者への曝露が既知である結論済みの試験から得られたデータのみを取り上げる。
表3 AURAa試験で報告された副作用の状況
| MedDRA 用語 | CIOMS 分類/ 全体頻度 (All) CIOMS 分類と全体的な頻度。 strong>CTCAE 分類)b |
3~4年生 | |
| 呼吸器.胸部および縦隔システムの疾患 | 間質性肺疾患c | 共通(2.7%)d。 |
0.7% |
| Gastrointestinal disorders | 下痢 | よくあること (42%) | 1% |
| 口内炎 | よくあること (12%) | 0% | |
| 皮膚および皮下組織障害 | ラッシュe | 非常に多い (41%) |
0.5% |
| 乾燥肌f | 非常に多い (31%) |
0% | |
| 爪真菌g | よくあること (25%) | 0% | |
| かゆみh | よくあること (14%) | 0% | |
| Laboratory tests (CTCAE Gradeの変更に伴い.テスト結果に基づき決定されます) 与えられた) | QT間隔の延長i | レア(0.2%) | |
| 血小板数の減少j | よくあること (54%) | 1.2% | |
| 白血球減少症j | よくあること (67%) | 1.2%。 |
|
| ノイトロペニアj | よくあること (33%) | 3.4% | |
。
a 表に示したデータは.AURA extension study(AURA ex; Phase II)およびAURA 2試験で得られた累積データであり.本剤を1回以上服用した患者さんに発生した有害事象のみをまとめています。
b 国立がん研究センター有害事象共通用語基準(NCI CTCAE)バージョン4.0。
c 次のカテゴリ用語が含まれる報告例:間質性肺炎.非感染性肺炎。
d CTCAEグレード5の事象(致死的事象)が4例報告されています。
e 発疹様事象のカテゴリー用語として.発疹.肉芽腫.紅斑.斑状発疹.丘疹.膿疱性発疹.紅斑.毛包炎.ざ瘡.皮膚炎.ざ瘡状皮膚炎が含まれる報告症例を対象としました。
f 乾燥肌.あかぎれ.乾燥性疾患.湿疹などの分類された用語の報告例を含む。
g 次のカテゴリー用語を含む報告例:爪床疾患.爪床の炎症.爪床圧痛.爪床変色.指(足)爪疾患.指(足)爪中毒.指(足)爪萎縮.指(足)爪感染.指(足)爪硬化.脆爪.爪剥離.脱色症.爪真菌症。
h 分類された用語:そう痒症.全身性そう痒症.眼瞼そう痒症の報告例を含んでいます。
i QTcFが500msec延長した患者を示す(心電図データから算出.報告された有害事象の発生率ではない)。
j は.報告された有害事象の発生率ではなく.実験室検査で見られた発生率を示しています。
AURA 17 安全性データ概要
アジア太平洋地域の171名(うち中国人患者148名)の前治療歴のあるT790M変異陽性NSCLC患者に対し.1日80mgの用量を投与した場合の安全性データが.アジア太平洋地域第2相試験(表4. AURA 17.【臨床試験】参照)で得られています。 AURA 17の安全性データは.グローバルな第II相安全性データと一致した。 有害事象の多くは.重症度グレード1または2でした。 最も多く報告された副作用は.下痢(29%)および発疹(20%)でした。AURA 17試験におけるCTCAEグレード3以上の有害事象の発生率は.14%でした。 本剤を1日80 mg投与された患者において.0.6%の患者が副作用により減量された。 1.2%の患者さんが副作用や臨床検査値異常により早期に投与を中止しています。
表 4. AURA 17 a 試験中に報告された副作用。
| MedDRA SOC | MedDRA 用語 | CIOMS 分類/ 全体頻度 (All) CIOMS 分類と全体的な頻度。 strong>CTCAE 分類)b |
3~4年生 |
| 呼吸器.胸部および縦隔システムの疾患 | 間質性肺疾患c | 共通(1.8%)d。 |
0% |
| Gastrointestinal disorders | 下痢 | よくあること (29%) | 0% |
| 口内炎 | 一般的(3.5%) | 0% | |
| 皮膚および皮下組織障害 | ラッシュe | 非常に多い (20%) |
0% |
| 乾燥肌f | 非常に多い (17%) |
0.6% | |
| 爪真菌g | 一般的(7.6%) | 0% | |
| 立ちくらみh | よくあること (13%) | 0% | |
| Laboratory tests (CTCAE Gradeの変更に伴い.テスト結果に基づき決定されます) 与えられた) | QTc interval prolongationi | 非常に稀 (0%) | |
| 血小板数の減少j | よくあること (65%) | 1.2% | |
| 白血球減少症j | よくあること (67%) | 0%。 |
|
| ノイトロペニアj | よくあること (29%) | 1.2% | |
。
a 表に示したデータは.AURA 17試験の最初のデータカットオフ日に基づくものです。 この時点では.すべての患者さんが18週間(4.5ヶ月)の治療を受ける機会がありました。本製品を少なくとも1回以上服用した患者さんに発生した有害事象のみをまとめています。
b 国立がん研究センター有害事象評価基準(Common Terminology Criteria for Adverse Events).バージョン4.0。
c 次の分類用語が含まれる報告例:間質性肺炎.非感染性肺炎。
d CTCAEグレード5の事象(致死的事象)が1例報告されています。
e 発疹様事象のカテゴリー用語として.発疹.肉芽腫.紅斑.斑状発疹.丘疹.膿疱性発疹.紅斑.毛包炎.ざ瘡.皮膚炎.ざ瘡状皮膚炎が含まれる報告例を対象としました。
f 乾燥肌.あかぎれ.乾燥性疾患.湿疹などの分類された用語の報告例を含む。
g 次のカテゴリー用語を含む報告例:爪床疾患.爪床の炎症.爪床圧痛.爪床変色.指(足)爪疾患.指(足)爪中毒.指(足)爪萎縮.指(足)爪感染.指(足)爪硬化.脆爪.爪剥離.脱色症.爪真菌症。
h 分類された用語:そう痒症.全身性そう痒症.眼瞼そう痒症の報告例を含んでいます。
i QTcFが500msecの患者を示す(心電図データから算出.報告された有害事象の発生率ではない)。
j は.報告された有害事象の発生率ではなく.実験室検査で見られた発生率を示しています。
特定の副作用の説明。
間質性肺疾患(ILD)。
第II相試験において.ILDは日本人の6.2%に発現し.非日本人アジア人患者の1.2%および非アジア人患者の2.4%に発現しました。ILDまたはILD様有害反応の発現までの期間の中央値は2.7カ月でした(【注意事項】を参照)。
QTcインターバルの延長。
AURAex試験及びAURA2試験の411例中.1例(0.2%)でQTc間隔が500msを超え.11例(2.7%)でQTc間隔がベースライン値から60ms以上延長した。 本剤の薬物動態解析では.濃度依存的にQTc間隔延長の発生率が増加することが予測されましたが.AURAex試験及びAURA2試験では不整脈の発生は報告されていません(【注意事項】を参照)。
心筋収縮率の変化。
AURAex試験およびAURA2試験(N=411)において.ベースラインおよび少なくとも1回のフォローアップ時のLVEF評価で.左室駆出率(LVEF)が>10%減少した患者は2.4%(9/375)であり.<50まで減少していることが示された。
高齢者の患者。
臨床試験期間中にAxitinibを服用した患者さん(N=411)のうち.46%が65歳以上.13%が75歳以上であった。 65歳以上の被験者では.65歳未満の被験者と比較して.試験薬の用量調節(投与停止または減量)に至る有害事象がより多く(23% vs. 17%)発生しました。 両カテゴリーの患者さん グレード3以上の副作用は.若年層と比較して高齢層でより多く発生しました(32%対28%)。
副作用が疑われる場合の報告。
医薬品が承認された後.疑わしい副作用を報告することが重要である。 これにより.製品のリスクとベネフィットのバランスを継続的に監視することができます。
【禁忌】。
有効成分または賦形剤に対して過敏症である。
本製品はセント・ジョーンズ・ワートと一緒に服用しないでください([薬物相互作用]を参照)。
【注意事項】。
EGFR T790M 変異状態の評価。
局所進行性・転移性NSCLCに対する本剤の使用を検討する際には.まずEGFR T790M変異の状態を明らかにする必要があります。 組織検体から採取した腫瘍DNAや血漿検体から採取した循環腫瘍DNA(ctDNA)は.十分に検証されたアッセイを使用して検査する必要があります。
腫瘍DNA中のT790M変異の状態を検査する場合(組織または血漿サンプルを介して).堅牢で信頼性が高く.感度の高いアッセイを使用しなければなりません。
組織または血漿検査による T790M 変異が陽性であれば.本製品による治療の適応となる。 しかし.血漿のctDNA検査で陰性となった場合.血漿検査では偽陰性の可能性があるため.可能であれば追加の組織検査を実施する必要がある。
間質性肺疾患(ILD)。
臨床試験において.本剤を使用した患者で重篤な.生命を脅かすまたは致死的な間質性肺疾患(ILD)またはILD様有害反応(非感染性肺炎等)が観察されています。 これらの事象の大部分は.本剤の投与中止後に改善または寛解します。 ILDの既往歴.薬剤性ILD.ステロイドホルモン療法を必要とする放射線肺炎.活動性ILDの臨床的証拠を有する患者は臨床試験から除外されました(【副作用】を参照)。
臨床試験において.間質性肺疾患(ILD)またはILD様有害事象(非感染性肺炎等)が本剤投与1,221例中2.9%に発現し.そのうち0.3%が死亡しました。 2つの第II相試験において.本製品を投与された411例中11例(2.7%)にILDまたはILD様有害事象が報告され.グレード3または4の有害事象は0.7%.死亡は1%に認められました。 試験期間中にILDが発生したのは.日本国籍の患者では6.2%であり.アジア国籍の患者では1.2%.非アジア国籍の患者では2.4%でした(【副作用】を参照)。
急性増悪及び/又は原因不明の肺症状(呼吸困難.咳.発熱)の増悪を示す患者を慎重に診察し.ILDを除外する。これらの症状の病因を探る間.本剤の投与を中止する。 ILDと診断された場合は.本剤の投与を永久に中止し.必要な治療措置を講じること。
QTcの延長間隔。
QTc間隔の延長は.心室性頻脈性不整脈(例:先端捻転型心室性頻脈)または突然死のリスクを高める可能性があります。AURAexまたはAURA2試験では不整脈イベントは報告されていません([有害事象]を参照)。 安静時心電図(ECG)検査により.心調律または心伝導に臨床的に重大な異常(例:QTc間隔>470ms)を有する患者を両試験から除外した([有害事象]を参照)。
先天性 QT 間隔延長症候群の患者には.可能であれば本製品を避けてください。 うっ血性心不全.電解質異常のある患者.またはQTc間隔を延長することが知られている薬剤を使用している患者は.心電図(ECG)と電解質の定期的な監視を受ける必要があります。 QTc 間隔 500ms を示唆する独立した心電図検査が 2 回以上ある患者については.QTc 間隔 481ms またはベースライン値(例:ベースライン QTc 間隔 481ms )に戻るまで一時的に投与を中止し.その時点で投与を再開できるが.表 1 に従って減量すること。 QTc間隔の延長と次のいずれかの症状が重なった場合.本剤の投与を永久に中止する必要があります:先端捻転型心室頻拍.多形性心室頻拍.重度の不整脈の徴候又は症状。
心筋収縮力の変化
AURAexおよびAURA2臨床試験において.ベースラインおよび少なくとも1回のフォローアップLVEF評価を行ったオセルチニブ投与患者の2.4%(9/375)にて.左室駆出率(LVEF)が>10%減少し.<50%まで減少しました。 入手可能な臨床試験データに基づくと.心筋収縮力の変化と本製品との因果関係は確立されていない。 心血管リスクが知られている患者やLVEFに影響を及ぼす可能性のある疾患を持つ患者では.ベースライン時および投与中のLVEF機能の測定を含む心機能のモニタリングを検討する必要がある。 本剤投与中に心イベントに関連する徴候や症状が現れた患者には.LVEF機能の測定を含む心臓モニターを検討する必要がある。
運転や機械操作の能力への影響。
本製品は.運転や機械操作の能力に影響を与えないか.または最小限の影響しか及ぼさない。
【妊娠中・授乳中の方へ】
女性の避妊と男性の避妊。
妊娠可能な年齢の女性は.本製品を服用中は妊娠を避けてください。 このような患者は.本製品の治療終了後.女性は少なくとも2ヶ月間.男性は4ヶ月間.有効な避妊を継続する必要があります。 本剤との併用により.ホルモン避妊薬の曝露量が減少するリスクを排除することはできない。
妊娠。
妊婦への使用に関するデータはないか.または非常に限られています。 動物実験では生殖毒性(胚死亡.胚発育遅延.新生児胎仔死亡.[薬理毒性]参照)が示唆されている。 作用機序及び前臨床試験データから.妊婦に使用した場合.胎児に有害である可能性がある。 本製品は.患者の臨床状態が本製品による治療を必要とする場合を除き.妊娠中には使用しないでください。
授乳について
本製品およびその代謝物がヒトの乳汁を介して排泄されるかどうかは不明である。 また.本製品またはその代謝物が動物の乳汁中に排泄されることを示唆する情報は不十分である。 しかし.授乳中の胎児から本剤及びその代謝物が検出され.胎児の成長及び生存に悪影響を及ぼすことがある(【薬理作用及び毒性】を参照)。 したがって.本製品が授乳中の乳児に影響を与える可能性は否定できない。 したがって.本剤投与中は授乳を中止すること。
妊産婦。
本製品がヒトの生殖機能に及ぼす影響についてのデータはない。 動物実験の結果.本剤は雌雄の生殖器に影響を与え.生殖能力を損なうことが示唆されている(【薬理作用】及び【毒性作用】参照)。
【小児用】。
歳未満の小児又は思春期の患者における本剤の安全性及び有効性は不明である。 これに関するデータはありません。
【老人用】。
臨床試験では.411例中187例(45%)が65歳以上.54例(13%)が75歳以上の患者さんでした。 年齢による効果の全体的な差は認められませんでした。 探索的解析の結果.65歳以上の患者さんではグレード3および4の副作用の発現率が高く(32% vs 25%).65歳未満の患者さんと比較して副作用による用量調節の頻度が高い(23% vs 17%)ことが示されました。
【薬物相互作用】。
ファーマコキネティック相互作用。
強力な CYP3A4 誘導物質により.本製品の曝露量が減少する可能性があります。 本製品は.BCRP基質への曝露を増加させる可能性があります。
オキシテトラサイクリンの血漿中濃度を増加させる可能性のある活性物質。
In vitro試験において.本製品は主にCYP3A4およびCYP3A5を介して代謝されることが確認されています(フェーズI)。 臨床薬物動態試験において.強力なCYP3A4阻害剤であるイトラコナゾール200 mg 1日2回投与との併用は.本剤の曝露量に臨床的に大きな影響を与えなかった(曲線下面積(AUC)が24%増加.Cmaxが20%減少した)。 したがって.CYP3A4阻害剤が本製品の曝露に影響を与えるとは考えにくい。 本製品の触媒となる他の酵素は同定されていない。
オセルチニブの血漿中濃度を低下させる活性物質。
臨床薬物動態試験において.リファンピシンとの併用投与(600 mg 1日1回.21日間)により.本剤の定常状態のAUCが78%減少した。 同様に代謝物であるAZ5104の曝露量も減少し.AUCとCmaxがそれぞれ82%と78%減少した。 本剤とCYP3A4の強力な誘導剤(フェニトイン.リファンピシン.カルバマゼピン等)との併用は避けることが望ましい。CYP3A4の中程度の誘導剤(ボセンタン.エファビレンツ.エトラビリン.モハフィニル等)も本剤の曝露量を減らす可能性があるので注意し.可能であれば併用しないようにすべきである。 CYP3Aの強力な誘導剤との併用が避けられない場合は.オキシチニブの用量を1日160mgに増量する必要があります。CYP3Aの強力な誘導剤の投与を中止してから3週間後に.オキシチニブの用量を1日80mgに戻してもよい。本製品とセントジョーンズワートの併用は禁忌(【禁忌】を参照)です。
酸分泌抑制剤によるoseltinibへの影響臨床薬物動態試験において.オメプラゾールの併用投与は本剤の曝露量に臨床的に適切な影響を与えなかった。 胃の中のpHを変化させる薬剤との併用も制限なく可能です。
オセルチニブ投与により血漿中濃度が変化する可能性のある他の活性物質。
in vitro試験の結果.本製品はBCRPトランスポータータンパク質に対して競争的阻害作用を有することが確認された。
臨床PK試験において.本剤とレスルバスタチン(感受性BCRP基質)の併用により.後者のAUCおよびCmaxはそれぞれ35%および72%増加しました。 BCRPに分布が依存し.治療域の狭い薬剤と本製品を併用する患者は.併用薬剤の曝露量の増加による忍容性の変化を適時に検出するために.注意深く観察する必要があります。
(薬物動態]参照)。
臨床PK試験において.本剤とCYP3A4感受性基質であるシンバスタチンとの併用により.後者のAUC及びCmaxがそれぞれ9%及び23%増加した。 この変化は小さいため.臨床的に重要であるとは考えにくい。 本剤は.PK の観点から CYP3A4 の基質と相互作用することはないと考えられる。 なお.CYP3A4以外のプレグナンX受容体(PXR)が調節する酵素との相互作用は調べていない。 本剤の併用により.ホルモン避妊薬の曝露量が減少する危険性を排除することはできない。
【薬物過剰摂取】。
第I/II相臨床試験において.少数の患者が用量制限毒性なしに240mgまでのoseltinibの1日投与量を受けたことがある。 これらの試験において.本剤の1日160mgおよび240mgの投与を受けた患者さんでは.80mg投与群と比較して.典型的なEGFR誘発性AE(主に下痢.発疹)の頻度と重症度の増加が認められました。 しかし.ヒトにおける偶発的な過剰摂取の経験はより限られています。 これらの症例はすべて.患者さんが誤って1回分を追加で服用した単発の事象であり.臨床的な影響はありませんでした。
本製品を過剰に摂取した場合の特別な治療法はありません。 過量投与が疑われる場合には.本剤の投与を中止し.対症療法を行うこと。
臨床試験
AURAexとAURA2のベースラインの特徴は.年齢中央値63歳.75歳以上13%.女性68%.白人36%.アジア人60%であった。 全患者が少なくとも1回の前治療を受けており.31%(N=129)が1回の前治療(EGFR-TKI治療のみ)を受け.69%(N=282)が2回以上の前治療を受けていた。患者の72%が喫煙経験があり.99%がWHO(世界保健機関)フィジカルステータススコア0または1.39%が脳 転移(少なくとも4週間安定で.副腎皮質ステロイド治療なし)。 大多数の患者(83%)はベースラインで既に内臓転移を有していた。追跡期間の中央値は.AURAex試験で6.9ヶ月.AURA2試験で6.7ヶ月であった。
AURA試験(第I相試験)は.オープンな単群用量漸増および延長第I相試験で.複数の用量漸増アームには.局所進行性または転移性のNSCLC患者271名が含まれています。 中央検査部での検査でEGFR T790Mが陽性であった治療患者63名の拡大コホートにおいて.80mg1日1回投与の有効性と安全性を検討した。 前治療にはEGFR-TKIと化学療法が含まれる。 このT790M陽性の研究集団(n=63)の人口統計学的特徴は.年齢中央値60歳.女性(62%).白人(35%).アジア人(59%).世界保健機関(WHO)の身体状況スコアが0または1の患者(100%).非喫煙者(67%)であった。 前治療のライン数は1~9本でした。 追跡期間中央値は8.2ヶ月。 表5は.AURA試験とそのプール解析(AURAexおよびAURA2)の有効性をまとめたものである。
表5.AURA試験の有効性結果。
| I 期間 | IIIssue | |||
| Efficacy indicators1 | AURA (Phase IExtended Cohort) (第I相)
。 |
AURAex (II期)
。 |
AURA 2 | Summary (N=411) |
| Objective Remission rate2,3. %(95% CI) |
62(48.74) | 61(54.68) | 71(64.77) | 66(61.71) |
| 寛解期間 (DoR)3 。 中央値.月(95%信頼区間) |
9.7(8.3.NE) | NE(NE.ネ) | 7.8(7.1.NE) | NE(8.3.NE) |
| 6ヶ月以上の病勢回復率(95%CI) | 72 (54,84) | 83(74.89) | 75(65.82) | 78(72.84) |
| Disease Control Rate(DCR)4. %(95% CI) |
95(86.99) | 90(85.94) | 91(87.95) | 91(88.94) |
| 無増悪生存期間。 中央値.月(95%CI) |
11(7.15) | NE(8.1.NE) | 8.6 (8.3, 9.7) | 9.7(8.3.NE) |
。
1 BICR(Blinded Independent Central Review)に基づき.PFSは経過観察中。
2寛解評価可能集団(ベースラインでBICRにより測定可能な病変を有する集団)に対してRECISTv1.1に基づきBICRにより決定した客観的寛解率.AURA.AURAex.AURA2.第II相試験のプール.n=60, 199, 199, 398; NE = not estimableであること。
3 寛解を経験した患者のみがカウントされました。DoRは.最初に記録された寛解(寛解は確認された完全または部分寛解と定義)後.疾患進行がない場合は記録された進行または死亡までの期間と定義されました。
4 病勢コントロール率は.完全寛解または部分寛解.あるいは6週間以上病勢が安定した患者の割合
客観的寛解率は.事前に定義されたすべてのサブグループ(治療ライン数.人種.年齢.地域)において50%を超えています。
全体では.1回目の画像検査(6週間)で86%(227/263例).2回目の画像検査(12週間)で96%(253/263例)が病勢寛解となった。
EGFR T790M de novo変異を有する患者を対象とした臨床試験は行われていない。
AURA17(n=171)は.アジア太平洋地域で診断され.EGFR感受性変異(EGFRm)およびEGFR T790M変異陽性の局所進行性または転移性非小細胞肺がん(NSCLC)患者において.既に治療を受けているAxitinib(80mg.1日1回経口投与)の第Ⅱ相オープン単群試験です。 承認済みのEGFR-TKI製剤による前治療後に病勢進行した患者さんにおける安全性と有効性の確認。 最近の治療時の病勢進行の出現と確認後.EGFR T790M変異の状態を中央検査室で検査できるように生検を行う必要があります(腫瘍組織のT790M変異の状態を調べるために.本試験ではRoche cobas® が使用されています)。 本試験における有効性の主要目的は.RECISTバージョン1.1による盲検独立施設審査(BICR)で評価された客観的寛解率(ORR)である。 副次的な有効性の目的は.寛解期間(DoR).疾患制御率(DCR).無増悪生存期間(PFS)を評価することでした。
AURA17の患者さんのベースライン特性は以下の通りです。本調査の患者さんの大多数は女性(117/171 [68.4%] 患者さん).アジア人(168/171 [98.2%] 患者さん).中国人(148/171 [86.5%] 患者さん)でありました。 試験参加時の年齢中央値は60.0歳(範囲:26-82歳).年齢層は50歳以上と65歳以上が最も多かった(79/171人[46.2%])。前治療(EGFR-TKI療法のみ)が1回の患者(N=54)が31.6%.2回以上を受けている患者(N=117)が68.4%であった。 の前治療を行った。 患者の大半は.転移性NSCLC(168/171 [98.2%]).組織型は腺癌(165/171 [96.5%]).WHO身体状態1(145/171 [84.8%])を有していた。 試験開始時の平均腫瘍負荷は.ベースライン時の総標的病変(TL)長径から66.1mm(sd.33.55)であり.大多数の患者はベースラインのTLサイズが40~79mmであった(77/171例[45.0%])。 患者の大半は内臓転移を有していた(141/171人[82.5%])。 追跡期間中央値は4.2ヶ月。 表 6 は.AURA17 試験の有効性をまとめたものである。
表 6 AURA17試験の有効性結果。
| Efficacy indicators1 | 全体. (N=171) |
中国サブグループ |
| 客観的寛解率2。 %(95% CI) |
60.2 (52.4, 67.7) | 59.7(51.2.67.8) |
| 89.1(79.0.94.5) | 89.9 (81.3, 94.7) | |
| 88.0 (82.0, 92.5) | 88.2 (81.8, 93.0) |
。
1PFSのフォローアップにおけるBICR(Blinded Independent Central Review)に基づく。
2寛解評価可能集団(ベースラインでBICRによる測定可能病変を有する集団)に対するRECISTv1.1に基づくBICRにより決定された客観的寛解率は.AURA17全体と中国人サブグループでそれぞれ166と144であった。
3 寛解を経験した患者のみがカウントされました。DoRは.最初に記録された寛解(寛解は確認された完全または部分寛解と定義)後.疾患進行がない場合は記録された進行または死亡までの期間と定義されました。
4 病勢コントロール率は.完全寛解または部分寛解.あるいは6週間以上病勢が安定している患者さんの割合です。
薬理学と毒性学]。
薬理作用。
オシチニブは.上皮成長因子受容体(EGFR)のキナーゼ阻害剤で.EGFRの特定の変異体(T790M.L858R.エクソン19欠失)に野生型の約9倍の低濃度で不可逆的に結合します。 細胞培養および動物移植腫瘍モデルにおいて.オシチニブはEGFR変異(T790M/L858R.L858R.T790M/エクソン19欠損.エクソン19欠損)を有する非小細胞肺がん細胞株に対して抗腫瘍活性を示し.野生型EGFR遺伝子増幅に対して弱い抗腫瘍活性を有していました。 オセルチニブの経口投与後.血漿中にオセルチニブと同様の阻害プロファイルを有する2つの薬理活性代謝物(AZ7550およびAZ5104.プロドラッグ化合物の約10%)が確認された。 AZ7550はオセルチニブと同様の効力を示し.AZ5104はEGFRエクソン19欠損およびT790M変異(約8倍)と野生型(約15倍)に高い効力を有していた。 より強い活動で In vitro試験で.オシチニブは臨床濃度において.HER2.HER3.HER4.ACK1およびBLK活性も阻害することが示されています。
毒性学的研究。
遺伝毒性: oseltinib Ames試験.マウスリンパ腫細胞試験及びラットin vivo小核試験の結果は全て陰性であった。
生殖毒性:動物実験では.オセルチニブは雄の動物で生殖能力を損なう可能性があることが示されています。 オシチニブを1ヶ月以上投与したラットおよびイヌでは.精巣に変性変化が見られ.ラットではその変化は可逆的であった。 オシチニブを40mg/kgの用量で約10週間投与したラットにおいて.未投与の雌ラットとヒト推奨用量(AUC80mg)の0.5倍を投与した雄との交尾後に産前産後の増加が認められ.雄における受胎能力の低下が示唆されました。
動物実験の結果に基づき.オキシチニブは雌の生殖能力を損なう可能性があります。 反復投与毒性試験の結果.オキシテトラサイクリンをヒト推奨用量80mgのAUCの0.3倍の曝露量で1カ月以上投与したラットでは.不活性化.卵巣の黄体の変性.子宮上皮及び膣上皮の菲薄化等の組織学的変化が観察されました。 投与1ヵ月後に観察された卵巣の変化は可逆的であった。 雌性生殖試験において.オシチニブは雌ラットに20mg/kg/日(ヒト推奨用量80mg/日の約1.5倍のCmax)を交配2週間前から妊娠8日目まで投与しても性周期や妊娠頭数には影響しないが.初期胚死亡を引き起こした。 雌ラットの交尾は.本剤投与中止後1ヶ月で可逆的であった。
ラット胚・胎児発生毒性試験において.オキシチニブ20mg/kg/日(血漿中曝露量は臨床曝露量の約1.5倍)を胚着床前から器官形成末期(妊娠2~20日)まで投与した妊娠ラットで着床後損失と初期胚死亡が認められた。 着床から硬口蓋閉鎖までの間(妊娠6~16日目)にオシチニブを1mg/kg/日以上(AUC値はヒト推奨用量80mgの0.1倍)投与した妊娠ラットにおいて.対照群と比較して投与群で胎児の奇形及び変動が増加する疑いがあることが示された。
周産期毒性試験において.オセルチニブ30mg/kg/日を投与した妊娠ラットの器官形成期から授乳期6日目までの全腹子の流産及び出生後の死亡率の増加が認められ.20mg/kg/日投与では出生時の平均仔体重のわずかな減少及び出生後の死亡率の増加が認められ.授乳期4~6日目から平均仔体重の増加が認められます。
発がん性:オセルチニブの発がん性に関する試験は実施されていない。
【薬物動態】。
本製品の薬物動態パラメータは.健常者とNSCLC患者を対象に特性評価を行いました。 母集団薬物動態解析に基づくと.見かけの血漿クリアランスは14.2 L/h.見かけの分布容積は986 L.終末半減期は約48時間であった。 また,20~240 mgの用量範囲では,AUCおよびCmaxは用量に比例していた。 Ocitinibは1日1回の経口投与で15日後に定常状態に達し.曝露量は約3倍に蓄積される。 定常状態では.循環血漿中濃度は24時間の投与間隔において通常1.6倍の範囲に維持されます。
吸収量。
オセルチニブの経口投与後.オセルチニブの血漿中濃度のピークはtmax(min-max)中央値で6(3-24)時間に到達しますが.投与後24時間の間に複数のピークを経験する患者もいます。 オキシチニブの絶対的なバイオアベイラビリティは測定されていない。 患者を対象とした80 mgの用量で実施した臨床薬物動態試験に基づき.食品は本剤のバイオアベイラビリティに臨床的に有意な影響を与えなかった。 (AUCは6%増加(90%CI -5,19)し,Cmaxは7%減少(90%CI -19,6)した)。 オメプラゾールを5日間投与し.胃内pHの上昇に伴い80 mg錠を投与した健康なボランティアにおいて.本剤の曝露量に有意な影響はなく(AUC及びCmaxはそれぞれ7%及び2%増加).曝露比の90%CIは80~125%の限界内であった。
配信オキシチニブの平均定常分布容積(Vss/F)は.母集団薬物動態モデリングにより986 Lと推定され.組織内における薬剤の幅広い分布が示唆されました。 血漿蛋白結合率は不安定なため測定できなかったが.製品の物理化学的性質から高い可能性がある。 また.ラットおよびヒト血漿タンパク質.ヒト血清アルブミン.ラットおよびヒト肝細胞と共有結合することが研究により確認されています。
生体内変換In vitro試験では.オキシチニブは主にCYP3A4およびCYP3A5を介して代謝されることが示唆されています。 このうち.CYP3A4を介した代謝は.二次的な経路であると考えられる。 さらに.in vitro試験では十分に定義されなかった他の代謝経路が存在する可能性があり.その後.オシチニブとともに経口投与した前臨床サンプルおよびヒト血漿から.2つの薬理活性代謝物(AZ7550およびAZ5104)が検出されました。AZ7550とオシチニブは同様の薬理特性を有しており.AZ5104は変異型および野生型のEGFRに対してより強力に作用することがわかっています。 投与後.これらの代謝物は血漿中にゆっくりと現れ.tmax(最小値-最大値)の中央値はそれぞれ24時間(4-72時間)および24時間(6-72時間)である。 ヒト血漿中では.原薬のオキシテトラサイクリンが全放射能の0.8%.上記の2つの代謝物がそれぞれ0.08%と0.07%を占め.放射能の大部分は血漿タンパク質に共有結合していた。 AUCに基づくと.定常状態におけるAZ5104およびAZ7550の曝露量の幾何平均は.それぞれoseltinibの曝露量の約10%であった。
オセルチニブの主な代謝経路は.酸化と脱アルキル化である。 ヒトの尿および糞便から少なくとも12成分が検出され.そのうち5成分が総投与量の1%以上を占めた。 このうち.製品の原型.AZ5104およびAZ7550はそれぞれ投与量の約1.9.6および2.7%を占め.システイン付加体(M21)と不明代謝物(M25)は約30%を占めた。 それぞれ1.5%.1.9%となりました。
In vitro試験において.オキシチニブはCYP3A4/5の競合阻害剤であるが.臨床的に意味のある濃度ではCYP1A2.2A6.2B6.2C8.2C9.2C19.2D6および2E1の阻害剤ではないことが示されました。 in vitro 試験に基づき.本製品は臨床的に有意な濃度では肝臓の UGT1A1 および UGT2B7 の阻害剤ではない。 また.腸内のUGT1A1を阻害する可能性があるが.臨床的に重要な影響を及ぼすかどうかは不明である。
消去法。
本剤20 mgを単回経口投与した場合,投与84日目の検体採取終了時までに,糞便から67.8%(試作品では1.2%),尿から14.2%(試作品では0.8%)が採取されました。 オシチニブプロトタイプは総排泄量の約2%を占め.尿と糞便からそれぞれ0.8%と1.2%が排泄されました。
輸送タンパク質との相互作用。
In vitroの研究では.オキシテトラサイクリンはOATP1B1およびOATP1B3の基質でないことが示されている。 また.in vitro試験において.本製品は臨床的に意味のある濃度ではOAT1.OAT3.OATP1B1.OATP1B3およびMATE2Kを阻害しないことが示されています。 しかし.MATE1やOCT2の基質との相互作用を排除することはできない。
オシチニブのP-gpとBCRPに対する効果について
In vitroの研究では.オキシチニブはP糖タンパク質および乳癌耐性タンパク質(BCRP)の基質であることが示されているが.臨床用量では.オキシチニブは関連活性物質との臨床的に意味のある薬物相互作用を引き起こす可能性は低い。 in vitro試験のデータから.oxitinibはBCRPおよびPgpの阻害剤であることが判明した。 しかし.CYP3A4以外のPXR制御酵素の相互作用は検討されていない([薬物相互作用]を参照)。
特殊な人口。
母集団薬物動態解析(n=778)において.定常状態の予測曝露量(AUCss)と以下の因子との間に臨床的に有意な関係は認められなかった:患者の年齢(範囲:21~89歳).性別.人種(白人.アジア人.日本人.中国人.非アジア系非白人含む).喫煙状況(現在喫煙者24名.禁煙者1名.喫煙者2名).喫煙量(AUCss)。 232). 母集団PK解析では.体重が意味のある共変量であることが示唆され.体重中央値(62kg)でのAUCssと比較して.90kgから43kgの範囲では.AxitinibのAUCssが-20%から+30%(95%から5%の四分位値)の変化となることが示されました。 体重の極値を考慮すると.代謝物AZ5104の比率の範囲は.<43kgから>90kgまで11.8%から9.6%.AZ7550の比率の範囲は12.8%から9.9%であった。 上記の体重差による曝露量の変化は.臨床的に有意なものではなかった。
肝機能障害。
オシチニブは主に肝臓から排泄されるため.本製品を服用する肝障害のある患者では曝露量が増加する可能性があります。 肝障害のある被験者での薬物動態試験は行われていない。 母集団PK解析に基づき.肝機能マーカー(ALT.AST.ビリルビン)とoxitinibの曝露量との間に有意な関係は認められなかった。 肝障害のマーカーである血清アルブミンは.oxitinibのPKに影響を及ぼした。 ASTまたはALTがULNの2.5倍.または悪性腫瘍に起因する場合はULNの5.0倍.総ビリルビンがULNの1.5倍の患者を除いて臨床試験が実施されています。 軽度の肝障害患者44名と肝機能正常患者330名の薬物動態解析に基づくと.両群の患者における曝露量は同程度であった。 肝障害のある患者における本製品の使用に関するデータは限られている([用法・用量]の項参照)。
腎臓障害。
腎障害のある被験者を対象とした薬物動態試験は行われていない。 の軽度腎機能障害(CLcr 60~<90 mL/min)患者330例.中等度腎機能障害(CLcr 30~<60 mL/min)患者149例.高度腎機能障害(CLcr 15~<30 mL/min)患者3例.正常腎機能(≥90 mL/min)患者295例に基づき.以下の通り算出されました。 これらの患者を対象とした母集団薬物動態解析では.oseltinib投与後の曝露量は同程度であった。 重度の腎機能障害は.肝臓を介した本剤の排泄に影響を与える可能性があります。 CLcrが15mL/min以下の患者は臨床試験の対象外とした。
人種。
AURA18試験(n=31)は.承認された市販のEGFR-TKIによる前治療(他の化学療法を併用.または併用しない)後に病勢進行した局所進行性または転移性NSCLCの中国人患者を対象とした第1相公開試験で.Oseltinibの2用量(40mgおよび80mg)の経口投与の薬物動態プロファイルを検討したものです。
オキシチニブの吸収は緩やかから中程度で持続性がありました。 オシチニブ(40mg~80mg)の単回及び複数回投与により.投与量にほぼ比例した曝露量の増加が認められた。 オセルチニブの見かけのクリアランスは低~中程度(単回投与で14.2L/時間.反復投与で15.3L/時間)で.広く分布していた(1113L)。
オシチニブは.単回投与後の半減期が約40時間で.15日間の投与で定常状態に到達します。 複数回投与後の定常状態(2サイクル目.1日目)では.曝露量の蓄積は約3.3倍であり.定常状態での薬物動態プロファイルは平坦であった。 2つの活性代謝物であるAZ5104とAZ7550は.定常状態ではオセルチニブと同様のフラットな薬物動態プロファイルを示し.それぞれ定常時のオセルチニブ曝露量の約12%から15%で循環しています。
中国人患者における経口オセルチニブの薬物動態プロファイルは.アジア人および非アジア人患者と同様であり.オセルチニブへの曝露は民族による影響を受けませんでした。
【ストレージ】。
30℃以下で保管してください。
【パッケージ】
アルミ製ダブルブリスター包装.1箱30錠(3プレート)入り。
ダブルアルミブリスターパック.1箱10錠(1プレート)入り。
[有効期限] .
36ヶ月。
[エグゼクティブスタンダード]。
輸入医薬品登録基準JX20160397。
[承認番号]。
[メーカー]。
会社名:AstraZeneca AB
生産拠点住所:Gärtunavägen, SE-151 85 Södertälje, Sweden
中国駐在員事務所住所:江蘇省無錫市新区黄山路2号
郵便番号:214028
品質苦情電話番号:400 828 1755.800 828 1755
製品情報フリーダイヤル:400 820 8116, 800 820 8116
ファックス: 021-38723255
ウェブサイト:www.astrazeneca.com.cn