承認日:
。
オラパリブ錠の使用方法
説明書をよく読み.医師の指導のもとでご使用ください
【薬剤名】
【薬剤名】
。
一般名:オラパリブ錠
販売名:Lipitor®/LYNPARZA®。
英語名:Olaparib Tablets
羽生ピンイン: アオラパリ・ピアン
[原材料名] 本製品の有効成分はOlaparibです。
化学名:4-(3-{[4-(シクロプロピルカルボニル)ピペラジン-1-イル]カルボニル}4-フルオロフェニル)メチル]フタラジン-1(2H)-オン
化学構造式。
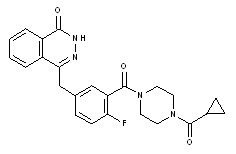
分子式: C24 H23 FN4 O3 。
分子量: 434.46
[プロパティ]本製品はフィルムコーティングされた錠剤です
150mg:緑色から緑色/灰色.楕円形の両凸錠で.片面に「OP150」の刻印があり.もう片面は空白である。
100mg:黄色~濃黄色の楕円形の両凸錠で.片面に「OP100」の刻印があり.もう片面は空白である。
【効能・効果】本剤は.白金製剤を含む化学療法による完全寛解または部分寛解後の.白金製剤感受性の再発卵巣がん.卵管がん.原発性腹膜がんの成人患者の維持療法に適応されます。
【スペック】(1) 150 mg.(2) 100 mg
【用法・用量】。
本製品は.抗悪性腫瘍剤の使用に経験豊かな医師の監督のもとで使用する必要があります。
推奨される投与量。
本製品は.150mgと100mgのサイズがあります。
なお.減量する場合は.100mg錠を使用する。
患者は.白金製剤を含む化学療法終了後8週間以内に本剤の投与を開始し.病勢進行または許容できない毒性が発現するまで治療を継続する必要があります。
投与方法。
経口投与。 本製品は丸ごと飲み込み.錠剤は噛んだり.砕いたり.溶かしたり.割ったりしないでください。 本製品は.食事と一緒に.または空腹時に摂取することができます。
投与ミス。
患者が薬の服用を忘れた場合.次の服用は通常通り予定時刻に行うこと。
投与量調節。
有害事象について
吐き気.嘔吐.下痢.貧血などの有害事象に対処するため.治療の中断や投与量の減量を検討します。
減量が必要な場合は.250mg(150mg錠1錠.100mg錠1錠)を1日2回(1日総量500mgに相当)に減量する。
さらに減量が必要な場合は.200mg(100mg×2錠)を1日2回(1日総量400mgに相当)に減量して服用する。
チトクロムP450(CYP) em>)3Aインヒビター。
本剤を使用する場合.強または中程度のCYP3A阻害剤との併用は推奨されず.他の代替薬を検討する必要があります。 強力なCYP3A阻害剤との併用が必要な場合は.本剤の用量を1回100mg(100mg錠1錠)を1日2回(1日総量200mgに相当)に減量することが推奨される。 中間作用型CYP3A阻害剤の併用が必要な場合は.150mg(150mg錠1錠)を1日2回(1日総量300mgに相当)に減量することが望ましい(【注意事項】及び【薬物相互作用】を参照)。
特殊な集団に対する薬。
腎臓障害:本剤は軽度の腎障害(クレアチニンクリアランス51~80mL/min)の患者には用量を調節することなく使用できる。中等度の腎障害(クレアチニンクリアランス31~50mL/min)の患者には本剤200mg(100mg錠2錠)1日2回(1日総量400mg相当)が推奨用量となる;重度の腎障害の患者には使用不可である 重度の腎障害又は末期腎不全(クレアチニンクリアランス≦30mL/min)の患者における本剤の安全性及び有効性に関するデータはなく.使用は推奨されない([薬物動態]の項参照)。
肝機能障害:。
本剤は軽度の肝障害(Child-Pugh 分類 A)の患者でも用量を調節することなく使用できる([薬物動態]の項参照)。 中等度又は重度の肝障害患者における本剤の安全性及び有効性に関するデータはなく.本剤の使用は推奨されない(【薬物動態】の項参照)。
小児または青少年:。
小児および青年に対する本剤の安全性および有効性は確立しておらず.小児患者への投与は推奨されない。
高齢者(>65歳):。
高齢者では開始時の用量調節は必要ありません。 75歳以上の患者さんについては.限られた臨床データしかありません。
【副作用】。
ある医薬品の臨床試験で観察された副作用の発現率は.他の医薬品の臨床試験で観察された副作用の発現率と直接比較することはできませんし.臨床試験は様々な異なる条件の下で実施されるため.臨床現場で観察される発現率を反映するものではありません。
再発卵巣がんに対する維持療法。
卵巣がん患者558名(オラパリブ投与331名.プラセボ投与227名)を対象に実施した臨床試験において.以下の副作用が報告されています。
SOLO-2。
SOLO-2試験では.プラチナ感受性生殖細胞由来乳がん感受性変異(gBRCAm)卵巣がん患者における本剤の維持療法としての安全性が評価されました。 本試験はプラセボ対照二重盲検試験で.294名の患者さんに.疾患進行または忍容できない毒性が発現するまで.本剤300mg(150mg×2錠)を1日2回(n=195)またはプラセボ錠を1日2回(n=99)投与しています。 試験期間中央値は.本製品投与群で19.4カ月.プラセボ投与群で5.6カ月でした。 グレードを問わず副作用による治療中止は.プラセボ群の18%に対し.本製品投与群では45%に認められ.副作用による減量は.プラセボ群の3%に対し.本製品投与群では27%に認められました。 治療の中断や減量に至った主な副作用は.貧血(22%).好中球減少(9%).疲労・脱力感(8%)などでした。 投与中止に至った患者さんの割合は.プラセボ群2%に対し.本製品群11%でした。
表1は.SOLO-2試験において本製品を投与された患者の20%以上に発現した有害事象をまとめたものです。 表2に.本剤を投与されたSOLO-2投与患者のうち.少なくとも25%に発現した臨床検査値異常を示す。
表1 SOLO-2における副作用a(≥20%) strong>オラパリブ錠で治療された患者。
副作用
オラパリブ錠
n=195
プラシーボ
n=99
1-4 学年.
%
Grade 3-4Grade 3-4 Grade 3-4Grade 3-4Grade 3-4Grade 3-4
%
第1学年から第4学年
.
%
第3-4学年.
%
。
血液およびリンパ系の疾患
貧血b
44
20
9
2
消化器系の疾患
.
吐き気
76
3
33
0
嘔吐
37
3
19
1
下痢
33
2
22
0
口腔粘膜炎c
口腔粘膜炎
20
1
16
0
感染症・伝染病
上咽頭炎/上気道炎/副鼻腔炎/鼻炎/インフルエンザ
上咽頭炎/上気道炎/鼻炎/インフルエンザ
36
0
29
0
全身性疾患.投与部位の各種反応
。
疲労感(脱力感を含む)
66
4
39
2
代謝障害および栄養障害
食欲不振
22
0
11
0
様々な筋骨格系および結合組織障害
関節痛/筋肉痛
30
0
28
0
あらゆる種類の神経障害
味覚障害
27
0
7
0
頭痛
26
1
14
0
。
。
a米国国立がん研究所有害事象評価基準(CTCAE)バージョン4.0に基づく評定。
bは.貧血.赤血球圧容量減少.ヘモグロビン減少.鉄欠乏症.平均細胞量増加.赤血球数減少などのカテゴリー項を表しています。
c は.口腔膿瘍.口内炎.歯肉膿瘍.歯肉疾患.歯肉痛.口腔潰瘍.粘膜感染.粘膜炎症.口腔カンジダ症.口腔不快感.口腔ヘルペス.口腔感染.口腔粘膜紅斑.口腔痛.中咽頭不快感.中咽頭痛などのカテゴリー用語のことです。
また.SOLO-2試験において本剤投与患者の20%に発現した副作用は.好中球減少.発疹.咳.消化不良.白血球減少.低マグネシウム血症.めまい.血小板減少.血清クレアチニン上昇.リンパ球減少及び浮腫であった。
表2 SOLO-2患者の25%以上で報告された臨床検査値異常
…….
Laboratory indicatorsa
オラパリブ錠
nb=195
Placebo
nb=99
1-4 学年。
%
第3-4学年
.
%
第1学年から第4学年
.
%
第3-4学年.
%
。
平均赤血球容積の増加c
89
–
52
–
ヘモグロビンの低下
83
17
69
0
白血球数の低下
69
5
48
1
リンパ球数の減少
67
11
37
1
絶対好中球数の減少
51
7
34
3
血清クレアチニン値上昇
44
0
29
0
血小板数の低下
42
2
22
1
。
。
a 臨床検査値がCTCAE grade 1の患者は.臨床試験への登録が許可されます。
b 安全性データセットの人数を表し.表中のデータは評価可能なすべての患者さんの各検査項目に基づいています。
c は.平均赤血球容積が正常上限値(ULN)である被験者の割合を表す。
勉強19。
オラパリブカプセルが投与された患者のうち.副作用による治療中断は35%(プラセボ群10%).減量が必要な患者は26%(同4%).中止となった患者は6%(同2%)であり.プラセボ群に比べ.副作用は少なかった。
表3は.19試験においてオラパリブ投与患者の20%以上に発現した有害事象をまとめたものです。 表4は.19試験でオラパリブを投与された患者の25%以上に発生した臨床検査値異常の一覧です。
表3 試験における副作用 19a 11
. strong>(オラパリブを投与された患者の20%以上)。
副作用
オラパリブカプセル.
n=136
プラシーボ
n=128
1-4 学年.
%
Grade 3-4Grade 3-4 Grade 3-4Grade 3-4Grade 3-4Grade 3-4
%
第1学年から第4学年
.
%
第3-4学年.
%
。
血液およびリンパ系の疾患
貧血b
23
7
7
1
消化器系の疾患
.
吐き気
71
2
36
0
嘔吐
35
2
14
1
下痢
28
2
25
2
便秘
22
1
12
0
全身性疾患.投与部位の各種反応
。
疲労感(脱力感を含む)
63
9
46
3
感染症・伝染病
呼吸器感染症
22
2
11
0
代謝障害および栄養障害
代謝障害および栄養障害
食欲不振
21
0
13
0
あらゆる種類の神経障害
頭痛
21
0
13
1
。
a 米国国立がん研究所(NCI)CTCAE 4.0分類による。
b 医学的な副作用の概念を反映した関連用語を表すカテゴリー用語。
また.19試験で本剤が投与された患者のうち20%に.消化不良.口腔粘膜炎.味覚異常.めまい.血中クレアチニン上昇.好中球減少.血小板減少.白血球減少.リンパ球減少.呼吸困難.発熱.浮腫等の副作用が認められました。
表4 試験参加者の25%以上が報告した検査項目 19 1. 検査異常。
Laboratory indicatorsa
オラパリブカプセル
nb=136
プラシーボ
nb=128
第1学年から第4学年
.
%
第3-4学年
.
%
第1学年から第4学年。
%
第3-4学年.
%
ヘモグロビンの減少
82
8インチ
58
1
平均赤血球容積の増加c
82
–
51
–
白血球数の低下
58
4
37
2
リンパ球数の減少
52
10
32
3
絶対好中球数の減少
47
7
40
2
血清クレアチニン値上昇
45
0
14
0
血小板数の低下
36
4
18
0
。
。
a 臨床検査値がCTCAE grade 1の患者は.臨床試験への登録が許可されます。
b 安全性データセットの人数を表し.表中のデータは評価可能なすべての患者さんの各検査項目に基づいています。
c は.平均赤血球容積が正常上限値(ULN)である被験者の割合を表す。
特定の副作用の説明。
血液学的毒性。
貧血やその他の血液毒性は全体的に低グレード(CTCAE グレード 1 または 2)でしたが.CTCAE グレード 3 以上の事象も報告されています。
貧血が初めて発現するまでの期間の中央値は約4週間(CTCAE≧グレード3の事象は約7週間)です。 貧血は.治療の中断および投与量の減量([用法・用量]を参照).および適切な場合には輸血によってコントロールすることができます。 SOLO2試験において.貧血の発生率は43.6%(CTCAE≧3では19.5%).貧血による投与中断.減量.中止の発生率はそれぞれ16.9%.8.2%.3.1%で.オラパリブ投与患者の17.9%が1回以上の輸血を必要としています。 オラパリブとヘモグロビン減少の間に量的効果関係が証明されています。 オラパリブ錠の臨床試験において.ベースラインに対するヘモグロビンの変化(低下)CTCAEグレード≧2の発生率は20%.好中球の絶対値は15%.血小板は5%.リンパ球は30%.白血球は20%(%はいずれも概数%)となっています。
平均赤血球容積がベースライン時の低値または正常値から正常上限値以上に増加した発現率は約55%であり.投与中止後に正常値に戻り.臨床的に重大な影響を及ぼすことはなかった。
治療開始後12ヶ月間は.ベースライン時の全血球数検査とその後の毎月のモニタリングが推奨され.その後は治療中に生じた臨床的に意味のあるパラメータの変化について定期的にモニタリングする。 これらの変化により.投与の中断または減量.さらなる治療が必要となる場合がある([用法]および[使用上の注意]を参照)。
その他の検査結果。
本製品の臨床試験において.血中クレアチニン値のベースラインからのCTCAE≧Grade 2の上昇の発生率は約15%でした。 プラセボ対照二重盲検比較試験のデータでは.血中クレアチニン値の中央値は投与前と比較して23%上昇し.投与中は維持され.投与中止後はベースラインに戻り.臨床的に重大な後遺症は認められませんでした。
吐き気と嘔吐。
吐き気は通常.治療のごく初期に発生し.ほとんどの患者さんで治療開始後1ヵ月以内に最初の吐き気が起こります。 嘔吐は通常.治療開始後早期に発生し.大多数の患者さんで治療開始後2ヶ月以内に最初の嘔吐が起こります。 吐き気および嘔吐は.大多数の患者で断続的であると報告されており.投与の中断.投与量の削減および/または制吐療法により制御することができます。 予防的制吐剤は必要ありません。
【禁忌】本剤の有効成分または賦形剤成分に対して過敏症のある人は禁忌である。 治療中および最終投与後1カ月間は授乳を中止させること([妊婦・授乳婦等への使用]の項参照)。
【注意事項】血液学的毒性。
本剤を投与された患者において.軽度または中等度(CTCAEグレード1または2)の貧血.好中球減少.血小板減少およびリンパ球減少の臨床診断および/または検査所見を含む血液学的毒性が報告されています。 患者は.以前の抗悪性腫瘍剤治療による血液学的毒性が回復するまで本剤の投与を開始しないこと(ヘモグロビン.血小板及び好中球のレベルがCTCAEグレード1以下まで回復すること)。 治療開始後12ヶ月間はベースライン時に全血球検査を行い.その後毎月モニタリングを行い.治療中に生じた臨床的に重要なパラメータの変化については.その後も定期的にモニタリングを行うことが推奨されます([副作用]を参照)。
重篤な血液毒性または輸血依存性の血液毒性が発現した場合は.治療を中断し.関連する血液学的検査を実施すること。 本剤の投与を4週間中断しても臨床的に異常な血液学的パラメータが残存する場合.骨髄検査及び/又は血液細胞遺伝学的検査を行うことが推奨される。
骨髄異形成症候群/急性骨髄性白血病と呼ばれるもの。
長期生存期間の追跡調査を含む臨床試験において.本剤単独投与患者における骨髄異形成症候群/急性骨髄性白血病(MDS/AML)の発現率は.<1.5%でした。 イベントの大半は.死亡という結果をもたらしました。 MDS/AMLを発症した患者において.オラパリブの投与期間は6ヶ月未満から2年以上と幅があり.より長い曝露期間に関するデータは限られています。 すべての患者はMDS/AMLの基礎因子を有し.白金製剤ベースの化学療法の前治療を受けていた。 一部の患者は.放射線治療だけでなく.DNAに損傷を与える他の薬物による治療も受けていた。 患者の大半はgBRCA 1/2変異のキャリアであった。 これらの患者さんの中には.過去に腫瘍や骨髄異形成の病歴がある方もいらっしゃいます。 オラパリブ錠の治療中にMDSおよび/またはAMLと診断された場合.オラパリブ錠の治療を中止し.適切な治療を行うことが推奨されます。
非感染性肺炎。
本剤単独投与時の臨床試験における非感染性肺炎(死亡に至る事象を含む)の発現率は1.0%でした。 報告された非感染性肺炎の臨床症状は様々で.多くの病因(肺がんおよび/または転移性肺がん.基礎疾患である肺疾患.喫煙歴および/または化学療法や放射線療法の既往歴)に影響されていた。 呼吸困難.咳嗽.発熱等の呼吸器症状が新たに発現又は悪化した場合.又は胸部画像所見が異常な場合は.治療を一時中断し.直ちに関連する検査を開始すること。 非感染性肺炎と診断された場合は.治療を中止し.適切な処置を行うこと。
胚性–胎児への毒性。
本剤の作用機序(ポリADPリボースポリメラーゼ阻害作用)及び動物実験から.妊婦に投与した場合.胎児に害を与える可能性がある。 ラットを用いた前臨床試験において.オラパリブの胚・胎児生存率への悪影響が示されています。 ヒトの推奨用量である1日2回300mg以下の曝露では.重度の胎児奇形が誘発される可能性があります。
本製品は.妊娠中は服用しないでください。 本剤服用中に患者が妊娠した場合には.胎児への危険性があることを患者に説明する必要がある。 妊娠可能な年齢の女性は.治療中および最終投与後6ヶ月間は効果的な避妊をすることが推奨されます。
男性患者及び妊娠可能な年齢の女性パートナーには.治療中及び最終投与後3カ月間は効果的な避妊を行い.精子提供を行わないよう助言すること([妊娠中及び授乳中の女性における使用]の項参照)。
他の医薬品との相互作用。
本剤と強又は中等度のCYP3A阻害剤との併用は推奨されない([用法・用量]を参照)。 強力または中程度のCYP3A阻害剤との併用が必要な場合は.投与量を減らす必要がある([用法・用量]を参照)。
本剤と強力または中間作用型の CYP3A 誘導剤との併用は推奨されない。 本剤の投与を既に受けている患者で.強力又は中間作用性CYP3A誘導剤による治療を必要とする場合.本剤の有効性が著しく低下する可能性があることを処方医は認識すべきである([薬物相互作用]の項参照)。
運転や機械操作の能力への影響。
オラパリブの運転や機械操作の能力への影響に関する研究は行われていません。 しかし.本剤投与中に脱力感.疲労感.めまいが報告されており.これらの症状が現れた患者さんは.運転や機械の操作に注意してください。
効果QTインターバル。
オラパリブの心臓再分極への影響は.300mg単回投与後の119名と300mg1日2回複数回投与後の109名で評価されました。 オラパリブのQT間隔に対する臨床的な関連性は認められませんでした。
[妊娠・授乳期について
【妊娠中・授乳中の方の使用】。
避妊。
妊娠可能な年齢の女性は.本製品による治療の開始時および治療中に妊娠してはならず.治療を開始する前に妊娠検査が必要です。 妊娠可能な女性は.治療中および本製品の最終投与後6カ月間は有効な避妊を行う必要があります([使用上の注意]を参照)。 オラパリブは酵素誘導によりCYP2C9基質への曝露を減少させる可能性があり.オラパリブとの併用によりホルモン避妊薬の効果が減少する可能性を否定できません。 したがって.治療中は他の非ホルモン性避妊手段を考慮し.定期的に妊娠検査を行う必要があります([薬物相互作用]の項参照)。
本剤の遺伝毒性試験及び動物生殖毒性試験に基づき.男性患者(配偶者が妊娠可能な年齢の女性又は妊婦)は.治療中及びオラパリブ最終投与後3カ月間は有効な避妊を行い.精子提供を行わないことが推奨されている(【注意事項】を参照)。
妊娠。
動物実験では.母体の全身曝露量が治療用量におけるヒトの曝露量よりも低いラット試験において.重度の催奇形性作用及び胚・胎児生存率への影響を含む生殖毒性が示されている([薬理学及び毒性]を参照)。 妊婦へのオラパリブの使用に関するデータはありませんが.オラパリブの作用機序に基づき.妊娠可能な年齢で確実な避妊を行っていない女性には.治療中および本製品の最終投与後6カ月間はオラパリブ錠を使用しないでください。
授乳。
オラパリブの母乳中への分泌に関する動物実験は実施されていない。 オラパリブ又はその代謝物がヒト母乳中に分泌されるかどうかは不明である。 本剤の薬理学的プロファイルに基づき.オラパリブ錠剤による治療中及び最終投与後1カ月間は授乳を中止することが望ましい([禁忌]を参照)。
妊産婦。
生殖能力に関する臨床データは得られていない。 動物実験では.被験薬は妊娠に影響を与えないが.胚・胎児の生存に悪影響を及ぼすことが示されている(【薬理作用と毒性】参照)。
【小児用】小児および青年に対する本剤の安全性および有効性は確立していないため.本剤は小児用としては不適当である。
【老人用】オラパリブ錠300mg1日2回単剤投与を受けた進行性固形癌患者687例を対象とした臨床試験において.146例(21%)が65歳以上.うち29例(4%)が75歳以上.85歳以上の患者さんはいませんでした。 オラパリブによる治療の安全性と有効性のデータには.若年層と高齢層の間で有意な差は見られませんでした。
[薬物相互作用
薬力学的相互作用。
本剤とDNAに損傷を与える薬剤を含む他の抗悪性腫瘍剤との併用による臨床試験では.骨髄抑制毒性のレベルの上昇と持続時間の延長が確認されています。 骨髄抑制作用を有する抗悪性腫瘍剤との併用には.単剤での推奨用量は適用されません。
オラパリブとワクチンや免疫抑制剤との併用試験は実施されていません。 したがって.上記の薬剤とオラパリブ錠の併用には注意が必要であり.患者の状態を十分に観察する必要があります。
ファーマコキネティック相互作用。
In vitro試験において.オラパリブはCYP3Aの阻害剤およびCYP2B6の誘導剤であることが確認されています。 オラパリブは.ヒトでは弱いCYP3A阻害剤であると予測されます。 In vitroの研究では.オラパリブが.ウリジン二リン酸グルクロニルトランスフェラーゼ(UGT)1A1.乳癌耐性タンパク質(BCRP).有機アニオン輸送タンパク質(OATP)1B1.有機カチオン輸送タンパク質(OCT)1.OCT2.有機アニオン輸送タンパク質(OAT)3.多剤・毒物排出輸送タンパク質(MATE)1およびMATE2Kの阻害剤であるかも示された。 の阻害剤である。 これらの知見の臨床的な関連性は不明である。 In vitro 試験において.オラパリブは排出トランスポーターである P-glycoprotein (P-gp) の基質であり.P-gp を阻害する。 オラパリブが P-gp を誘導する可能性は評価されていない。
オラパリブの血漿中濃度を上昇させる可能性がある薬剤。
オラパリブは.主にCYP3Aによって代謝されます。 患者(n=57)において.強力なCYP3A阻害剤であるイトラコナゾールの併用により.オラパリブのAUCが170%増加しました。 中活性型CYP3A阻害剤フルコナゾールは.オラパリブの血漿中濃度対時間曲線下面積(AUC)を121%増加させると予測された。
イトラコナゾール.テリスロマイシン.クラリスロマイシン.ケトコナゾール.ボリコナゾール.ネファゾドン.ポサコナゾール.リトナビル.ロピナビル/リトナビル.インジナビル.サキナビル.ネルフィナビル.ボセプレビル.テラプレビル等の強力なCYP3A阻害剤やアンプレナビル等のCYPA中間阻害剤の併用は避け.また.これらの阻害剤と併用することで.より高い効果が期待できます。 (アンプレナビル).アリピタント.アタザナビル.シプロフロキサシン.クリゾチニブ.ダルナビル/リトナビル.ジルチアゼム.エリスロマイシン.フルコナゾール.ホスアンプレナビル.イマチニブ.ベラパミル。 強または中程度のCYP3A阻害剤の併用が必要な場合は.オラパリブの投与量を減量する必要があります。
グレープフルーツ.グレープフルーツジュース.ライム.ライムジュースはCYP3A阻害剤を含むため.オラパリブ治療中は避けてください。
オラパリブの血漿中濃度を低下させる可能性のある薬剤。
患者(n=22)では.強力なCYP3A誘導物質であるリファンピシンの併用により.オラパリブのAUCが87%減少しました。 中活性型CYP3A誘導剤は.オラパリブのAUCを約60%減少させると予測された。
フェニトイン.リファンピシン.カルバマゼピン.セントジョーンズワートなどの強力なCYP3A誘導剤).ボセンタン.エファビレンツ.エトラビリン.モダフィニル.ナフシリンなどの中間作用型CYP3A4誘導剤の併用は避けてください。 中間作用型CYP3A誘導剤の使用を回避できない場合.オラパリブの有効性が低下する可能性があります。
【薬物過剰摂取】本製品による過量投与の臨床的徴候は確認されていない。 オラパリブ錠を1日900mgまで2日間服用した少数の患者において.予期せぬ副作用は報告されていません。 過量投与に対する特別な管理はありません。 過量投与が発生した場合.医師は対症療法および一般的な支持療法を行う必要があります。
【臨床試験】再発卵巣癌に対する維持療法。
プラチナ製剤を含む治療で寛解を得た再発卵巣癌患者を対象とした2つの無作為化プラセボ対照二重盲検多施設共同試験で.olaparibの有効性が評価されました。
SOLO2研究 (D0816C00002) 研究 () 研究 () 研究 ()
SOLO2試験は.gBRCA1/2変異を有するプラチナ感受性再発卵巣がん.卵管がん.原発性腹膜がんの患者さんにおける維持療法としてのオラパリブの安全性と有効性を評価した無作為化二重盲検プラセボ対照第III相試験です。 白金製剤を含む化学療法終了後に寛解(完全寛解[CR]または部分寛解[PR])した高悪性度漿液性または内膜性PSR卵巣がん患者295名を登録し.2対1の割合で(オラパリブ群196名.プラセボ群99名).病勢進行または以下の症状が発現するまでオラパリブ(300mg[150mg錠2]1日2回)またはプラセボの投与を行いました。 耐容性のない毒性
2種類以上の白金製剤を含む化学療法を受けた患者は.最後の白金製剤を含む化学療法終了後6ヶ月以内に疾患が再発するまで研究に参加しました。 オラパリブまたは他のPARP阻害剤による前治療を受けていない患者であること。 ベバシズマブによる前治療歴は認められますが.無作為化前の治療レジメンにベバシズマブが含まれていないことが条件となります。
ベースライン時.すべての患者が.局所検査(n=236)または中央Myriad CLIA(n=59)で.その後BRACAnalysis® CDx(n=286)で確認したgBRCA 変異を有しているか.または変異が疑われる患者。 無作為化された患者の4.7%(14/295人)でBRCA1/2遺伝子にラージフラグメント再配列が検出された。
人口統計学的特性およびベースラインの特性は.オラパリブ群とプラセボ群でほぼ同じでした。 両群の年齢の中央値は56歳であった。 卵巣がんが80%以上の患者さんの原発であった。 最も多い組織型はplasmacytic (> 90%)で.6%の患者にはendometrioid carcinomaが見られた。 オラパリブ群では.過去に二次治療のみを受けていた患者さんが55%.三次治療以上を受けていた患者さんが45%でした。 プラセボ群では.61%の患者さんが二次治療のみ.39%の患者さんが三次治療以上の治療を受けていました。 白金製剤を含む化学療法への反応性は.完全寛解が47%.部分寛解が53%であり.ECOG fitness scoreは0が81%と大半の症例で認められた。 オラパリブ群とプラセボ群では.それぞれ17%と20%の患者さんがベバシズマブによる前治療を受けていました。
主要評価項目は無増悪生存期間(PFS)で.RECIST(Remission Evaluation Criteria in Solid Tumours)1.1評価法を用いて治験医師により測定されました。 副次的評価項目は.二次疾患進行または死亡までの期間(PFS2).OS(全生存期間).治療中止または死亡までの期間(TDT).最初の抗がん剤治療開始または死亡までの期間(TFST).二次抗がん剤治療開始または死亡までの期間(TSST).HRQoL(健康に関する生活の質)とし.これらの評価項目が達成されたことを確認した上で.本試験の結果を発表しました。 ).
本試験では.主要評価項目であるPFSがプラセボ群に対してオラパリブ群で統計的に有意に改善し.リスク比(HR)は0.30(95%CI 0.22-0.41; p<0.0001; 中央値はオラパリブ群19.1カ月に対してプラセボ群5.5カ月)であった。 治験責任医師が評価したPFSの結果は.盲検化された独立した中央画像評価によって裏付けられました(HR=0.25.95% CI 0.18-0.35.p<=0.0001.中央値はオラパリブ群30.2カ月.プラセボ群5.5カ月)。2年後.プラセボ群と比較してオラパリブ投与群の43%が無進行を維持しました。 は15%にとどまりました。
SOLO2におけるgBRCA1/2m PSR卵巣がん患者様の主要評価項目の結果概要については.表5および図1をご覧ください。
表5 SOLO2におけるgBRCA1/2m PSR 卵巣癌患者の主要評価項目。 試験結果の概要(治験責任医師による評価)。
Olaparib300 mg錠。
(n=196)
プラシーボ
(n=99)
PFS(63%) 成熟度
イベント数:総患者数(%)
107:196(55)
80:99 (81)
期間中央値(月)(95%CI)
19.1(16.3〜25.7)
5.5(5.2-5.8)
HR (95% CI)a
0.30 (0.22-0.41)
P値(両側)
p<0.0001
。
。
a HR=危険率。 Value<1 はオラパリブを支持する。 白金製剤を含む前治療の化学療法における寛解反応(CRまたはPR)および病勢進行までの期間(>6-12ヶ月および>12ヶ月)で層別化し.log-rank検定を用いて解析を行った。
bd 1日2回投与;PFS 無増悪生存期間;CI 信頼区間。
図1 SOLO2: gBRCA1/2m PSR 卵巣癌患者のPFSのKaplan-Meier曲線(63%)。 >成熟度 – 調査員評価)。
ランダム化以降の期間(月)
。
プラシーボ
。
。
Olaparib300 mg 1日2回
。
。
危険にさらされている患者数
。
。
プラシーボ
。
。
Olaparib300 mg 1日2回
。
。
bd 1日2回; PFS 無増悪生存期間
副次評価項目であるTFSTとPFS2は.プラセボ群と比較してオラパリブ群で持続的かつ統計的に有意な改善が認められました(表6)。
表6 SOLO2gBRCA1/2m PSR 患者における主要な二次試験について。 エンドポイント結果の概要
Olaparib300 mg錠。
(n=196)
プラシーボ
(n=99)
TFST (58% maturity)
インシデント数:総患者数(%)
92:196 (47)
79:99(80)
期間中央値(月)(95%CI)
27.9(22.6-NR)
7.1(6.3-8.3)
HR(95%信頼区間)a
0.28(0.21-0.38)。
P値*(バイラテラル)
p<0.0001
PFS2(40%) 成熟度
インシデント数:総患者数(%)
70:196 (36)
49:99 (50)
期間中央値(月)(95%CI)
NR(24.1-NR)
18.4(15.4-22.8)
HR(95%信頼区間)a
0.50(0.34-0.72)。
P値(両側)
p=0.0002
。
。
* 制御不能な多重性
a HR=リスク比。 値 <1 はオラパリブを支持する。 白金製剤を含む前治療の化学療法における寛解反応(CRまたはPR)および病勢進行までの期間(6-12ヶ月および12ヶ月)で層別化し.log-rank検定を用いて解析した。
bd 1日2回投与.達成度はNR.CI信頼区間.PFS2は2回目の病勢進行または死亡までの時間.TFSTは最初の追跡治療開始または死亡までの時間。
本試験に登録された測定可能病変(ベースライン時の標的病変)を有する患者さんのうち.客観的寛解率は.プラセボ群17%に対し.オラパリブ錠群41%でありました。 試験開始時に病変(ベースライン時の標的病変または非標的病変)を有していたオラパリブ錠剤投与患者の15.0%で完全寛解が得られたのに対し.プラセボ投与患者では9.1%であった。
患者さんの自己報告による転帰(PRO)データでは.ベースラインの変化に対するFACT-O(Functional Assessment of Cancer Therapy-Ovarian Cancer)試験の転帰指標(TOI)で評価すると.オラパリブ群とプラセボ群との間に差は見られませんでした。
SOLO2研究 (D0816C00002)
研究 () 研究 () strong>–中国コーホート。
SOLO2試験には.中国人患者さんが別のコホートとして登録され.合計32名の中国人患者さんが.病勢進行または忍容できない毒性が発現するまで.300mg(150mg×2錠)を1日2回投与(n=22)またはプラセボ錠を1日2回投与(n=10)のいずれかにランダムに割り振られました。 中国人の被験者は深センのUW Clinical Laboratoryで検査を受けてgBRCA変異があることが確認された。 本製品を投与された患者さんの年齢中央値は49歳(範囲:37~65歳).プラセボを投与された患者さんの年齢中央値は46.5歳(範囲:33~67歳)となっています。 ECOGフィジカル・ステータス・スコアは.治療群で73%.プラセボ群で50%が0でした。全患者の59%が無作為化前の最後の白金製剤を含む化学療法後に完全寛解を達成し.56%の患者は最後の白金製剤を含む化学療法完了後6〜12カ月で病勢進行までの時間を達成しました。 オラパリブ群の約23%.プラセボ群の約20%が.3ライン以上の白金製剤を含む前治療を受けていました。 SOLO2 Chinaのコホートにおいて.本製品はプラセボと比較して.患者さんのPFS(治験担当医師による評価)をHR 0.44(95%CI:0.17-1.19.p=0.0776.中央値:オラパリブ群13.8ヶ月.プラセボ群5.5ヶ月)と.有意に改善させました。 この結果は.独自に行った画像評価の結果とも一致します。 患者の生存率に関するデータはまだ成熟していない(19%の患者にのみイベントが発生した)。
表7 SOLO2Chinese CohortgBRCA1/2mPSR 卵巣癌の患者さん 主要評価項目(治験責任医師評価)の結果のまとめ。
。
オラパリブ錠 300 mg 錠 1日2回投与!
(n=22)
プラシーボ
(n=10)
PFS(65.6%) 成熟度
イベント数:総患者数(%)
14:22 (63.6)
7分10秒(70.0)
期間中央値(月)(95%CI)
13.8 (10.1, 16.6)
5.5(3.2.8.4)
リスク比 HR (95% CI)a
0.44 (0.17, 1.19)
P値(bilateral)a
p=0.0776
。
。
a HR=リスク比.リスク比対p値は非階層化比例リスクモデルによるもの。
PFS 無増悪生存期間
中国人を対象とした有効性および安全性は.非中国人を対象とした有効性および安全性と同様であった。
Study19(D0810C00019Study 19 em>)。
19試験は.プラチナ製剤による2種類以上のレジメン投与後のPSR卵巣がん患者(卵管がん.原発性腹膜がんを含む)を対象に.維持療法としてのオラパリブの安全性と有効性を評価する無作為化二重盲検プラセボ対照の第II相試験です。 白金製剤を含む化学療法終了後に寛解(CRまたはPR)を得たPSRグレードのプラズマ卵巣がん患者265名を登録し.1:1で無作為化(オラパリブ群136名.プラセボ群129名)し.疾患進行または忍容できない毒性が発現するまでオラパリブのカプセル400mg(50mg×8錠)1日2回(中国ではカプセルの入手は申告されていません)とプラセボの投与を実施しました。 主要評価項目はRECIST 1.0基準を用いて治験責任医師が評価したPFSで.副次的評価項目はOS.疾患制御率(DCR.すなわちCR/PR+SD(疾患安定化)確認).HRQoL.疾患関連症状などでした。 TFSTとTSSTについては.探索的な解析を行った。
本試験では.白金製剤を含む最後の化学療法終了から疾患再発までの6ヶ月間の患者を登録しました。 組み入れ条件には.BRCA1/2変異の同定は含まれなかった(一部の患者はBRCA変異の状態についてレトロスペクティブに検査されている)。 オラパリブまたは他のPARP阻害剤による前治療を受けていない患者であること。 ベバシズマブによる治療歴があってもよいが.無作為化前の治療レジメンにベバシズマブが含まれていないことが条件とされた。 オラパリブ治療中に病勢進行が生じた場合.それ以降のオラパリブによる治療はできません。
BRCA1/2変異を有する患者は.局所検査または血液生殖細胞検査用のMyriad CLIA comprehensive BRACAnalysis® 検査.あるいはFoundation Medicineが実施する検査を用いて腫瘍サンプルで行われる検査を用いて同定されました。 無作為化された患者の7.4%(10/136人)でBRCA1/2遺伝子にラージフラグメント再配列が検出された。
人口統計学的特性およびベースライン特性は.オラパリブ群とプラセボ群で基本的に類似していました。 両群の年齢の中央値は59歳で.86%が原発性卵巣癌であった。 オラパリブ群では.過去に二次治療のみを受けていた患者さんが44%.三次治療以上を受けていた患者さんが56%でした。 プラセボ群では.49%の患者さんが二次治療のみ.51%の患者さんが三次治療以上の治療を受けていました。 白金製剤を含む化学療法に反応した患者は.45%が完全寛解.55%が部分寛解であった。 オラパリブ群およびプラセボ群では.ベバシズマブによる前治療を受けた患者はそれぞれ6%と5%でした。
本試験では.主要評価項目であるPFSがプラセボ群に比べオラパリブ群で統計的に有意に改善し.HR 0.35 (95% CI 0.25-0.49; p<0.00001; 中央値はオラパリブ群 8.4 ヶ月 対 プラセボ群 4.8 ヶ月)となりました。 最終的なOS解析(成熟度79%.データカットオフ日[DCO]2016/05/09)では.オラパリブとプラセボのリスク比は0.73(95%CI 0.55-0.95;p=0.02138[specified significance level not reached<;0.0095]; 中央値はオラパリブ群では29.8ヶ月.プラセボ群では (27.8ヵ月)。 オラパリブ投与群では.23.5%(n=32/136)の患者が2年以上の治療を受けたのに対し.プラセボ投与群では3.9%(n=5/128)の患者が2年以上の治療を受けていました。 患者数が限られているにもかかわらず.オラパリブ群の13.2%(n=18/136)が5年以上治療を受けたのに対し.プラセボ群では0.8%(n=1/128)でした。
事前に計画されたサブグループ解析の結果.卵巣がんのBRCA1/2変異を有する患者さん(n=136, 51.3%; 体細胞BRCA1/2変異が確認された20名を含む)には.olaparib単独療法の維持療法の効果が最も高いことがわかりました。 また.BRCA1/2野生型/意義不明の変異体(BRCA1/2 wt/VUS) 患者においても.より少ないながらも臨床的有用性が観察されました。 サブグループ解析のための多重検定ができなかった。
19試験におけるBRCA1/2変異およびBRCA1/2 wt/VUS PSR卵巣がん患者における主要試験評価項目の結果の概要は.表8と図2をご覧ください。
表8 研究 19BRCA1/2
strong>変異とBRCA1/2 wt/VUS PSR 卵巣癌患者における主要評価項目の結果概要
卵巣癌患者における主要評価項目の結果は.次のとおりです。
All patientsa
BRCA1/2mutations
BRCA1/2 WT/VUS
Olaparib400 mg.
カプセルbd
プラシーボ
Olaparib400 mg.
カプセルbd
プラシーボ
Olaparib400 mg.
カプセルbd
プラシーボ
PFS-DCO 2010年06月 302010
strong>日
イベント数:総患者数
(%)
60:136
(44)
94:129
(73)
26:74
(35)
46:62
(74)
32:57
(56)
44:61
(72)
期間中央値(月)(95%CI)
8.4
(7.4-11.5)
4.8
(4.0-5.5)
11.2
(8.3-NR)
4.3
(3.0-5.4)
7.4
(5.5-10.3)
5.5
(3.7-5.6)
HR (95% CI)b
0.35(0.25〜0.49)
0.18(0.10〜0.31)
0.54(0.34〜0.85)
P値(両側)
p<0.00001
p<0.00001
p=0.00745
。
。
a 以下のサブグループを含む全患者: BRCA1/2-mutation, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with unknown states not included in the
表は.独立したサブグループとしてではなく)。
b HR=リスク比。 値 <1 はオラパリブを支持する。 解析は.治療法.民族的起源.プラチナ感受性についてCox比例リスクモデルを用いて行われた。
と最後の白金製剤ベースの化学療法後の寛解をエフェクターとして使用した。
wt(野生型)野生型;VUS(意義不明の変異)意義不明の変異;bd 1日2回;PFS 無増悪生存期間;DCOデータカットオフ日;CI 信頼区間;NR 未達成。
図219試験:FASにおけるPFSのKaplan Meier カーブ(58%):FASにおけるPFS カーブ。 strong>Mature –Researcher Assessment) DCO 2010 06 30 6月 Mature –Multi –Multi –
ランダム化以降の期間(月)
。
プラシーボ
。
。
Olaparib400mg
bd>。
。
Olaparib400mg
bd>。
。
プラシーボ
。
。
危険にさらされている患者数:
。
。
bd 1日2回; DCO data cut-off date; FAS full analysis set; PFS progression-free survival
19試験におけるBRCA1/2変異およびBRCA1/2 wt/VUS PSR卵巣がん患者さんの主要副次評価項目の結果を表9に.全患者さんの結果を図3にまとめました。
表9 研究 19BRCA1 >/2変異とBRCA1/2 wt/VUS PSR 卵巣癌患者における主要副次評価項目の結果概要 卵巣癌患者における主要副次評価項目の結果概要。 強い>。
All patientsa
BRCA1/2mutations
BRCA1/2 wt/VUS
BRCA1/2BRCA1/2
Olaparib400 mg.
カプセルbd
プラシーボ
Olaparib400 mg.
カプセルbd
プラシーボ
Olaparib400 mg.
カプセルbd
プラシーボ
。
OS – DCO 2016Year05Month09Year05Month strong>日
インシデント数:総患者数(%)
98:136
(72)
112:129
(87)
49:74
(66)
50:62
(81)c
45:57
(79)
57:61
(93)
平均時間(月)
(95% CI)
29.8
(26.9-35.7)
27.8
(24.9-33.7)
34.9
(29.2-54.6)
30.2
(23.1-40.7)
24.5
(19.8-35.0)
26.6
(23.1-32.5)
HR (95% CI)b
0.73(0.55〜0.95)
0.62(0.42〜0.93)
0.84(0.57〜1.25)
P値*(バイラテラル)
p=0.02138
p=0.02140
p=0.39749
TFST – DCO 2016Year05Month09Year05Month0strong>Year/No. strong>日
インシデント数:総患者数(%)
106:136
(78)
124:128
(97)
55:74
(74)
59:62
(95)
47分57秒
(83)
60:61
(98)
平均時間(月)
(95% CI)
13.3
(11.3-15.7)
6.7
(5.7~8.2)
15.6
(11.9-28.2)
6.2
(5.3-9.2)
12.9
(7.8-15.3)
6.9
(5.7-9.3)
HR (95% CI)b
0.39(0.30〜0.52)
0.33(0.22〜0.49)
0.45(0.30〜0.66)
P値*(バイラテラル)
p<0.00001
p<0.00001
p=0.00006
。
。
* サブグループ解析や全患者のTFSTに多重検定戦略はなかった。
aすべての患者さんには以下のサブグループが含まれます:BRCA1/2変異.BRCA1/2 wt/VUS および BRCA1/2 状態不明(11名は.BRCA1の状態不明)。
表は.独立したサブグループとしてではなく)。
b HR=リスク比。 値 <1 はオラパリブを支持する。 解析は.Cox比例リスクモデルを用いて.治療法.人種.プラチナ感受性.および
白金製剤を用いた最終化学療法後の寛解をエフェクターとする。
cBRCA変異サブグループでは.プラセボ投与群の約4分の1(14/62;22.6%)が.その後PARP阻害剤による治療を受けました。
wt(野生型)野生型;VUS(意義不明の変異)意義不明の変異;bd 1日2回;OS 全生存期間;DCO データカットオフ;CI:信頼区間;TFST ランダム化から最初の後続治療または死亡までの時間
<図3 試験19:FASにおけるOS(79%)のカプラン・マイヤー曲線。 strong>Maturity, DCO 2016 May 09) Maturity, DCO 2016
プラシーボ
。
。
Olaparib400mg
bd>。
。
Olaparib400mg
bd>。
。
Placebo
。
危険にさらされている患者数:
。
ランダム化以降の期間(月)
。
。
bd 1日2回; DCO data cut-off date; FAS full analysis set; OS overall survival
患者さんの自己報告による転帰(PRO)データでは.Functional Assessment of Cancer Therapy – Total Ovarian Cancer Score(FACT-O)のTrial Outcome Index(TOI)で測定した改善率および悪化率により.olaparib群とプラセボ群に差がないことを示しました。
【薬理学・毒性学】
薬理作用。
Olaparibは.ポリADPリボースポリメラーゼ(PARP.PARP1.PARP2.PARP3)阻害剤であり.PARPは.DNA転写やDNA修復などの正常な細胞機能に関与している。 オラパリブは.in vitroでは腫瘍細胞株の増殖を.in vivoではヒト腫瘍搭載マウス異種移植片の増殖を抑制し.単剤またはプラチナ製剤による化学療法後の投与で有効であることが試験で示されています。 オラパリブ投与により.白金系化学療法反応に伴うDNA損傷相同組換え修復のBRCA関連欠損.または非BRCA関連欠損を有する細胞株およびマウス移植腫瘍モデルにおいて細胞障害および腫瘍抑制作用が増強されました。 In vitroの研究では.オラパリブの細胞毒性は.PARPase活性の阻害とPARP-DNA複合体形成の増大が関与し.DNA損傷とがん細胞死を引き起こすことが示唆されています。
毒性試験。
遺伝毒性: オラパリブのエームス試験の結果は陰性でした。中国ハムスター卵巣(CHO)細胞染色体異常試験およびラット骨髄小核試験の結果は染色体切断作用が陽性で.オラパリブの薬理作用と一致しました。 ゲノムの不安定性はオラパリブの薬理作用と一致し.オラパリブがヒトにおいて遺伝毒性を有する可能性が示唆された。
<生殖毒性:雌ラットの生殖能力及び初期胚発生毒性試験において.交配14日前から妊娠6日目までオラパリブを最大15mg/kg/日(母親の全身曝露量は臨床推奨ヒト曝露量[AUC0-24h]の約7%)で経口投与したところ.以下の影響は認められませんでした。 交配および生殖能力への影響は見られなかったが.生殖後の流産が増加した。
雄ラットの生殖能力試験において.オラパリブを40mg/kg/日までの用量(全身曝露量は臨床推奨ヒト曝露量[AUC0-24h]の約5%)で少なくとも70日間経口投与したところ.交尾及び生殖能力に対する影響は認められなかった。
胚・胎児発生毒性試験において.器官形成期の妊娠ラットにオラパリブ 0.05 および 0.5 mg/kg/day を経口投与した。 胚・胎児毒性は0.5 mg/kg/日の用量(母親の全身曝露量はヒトの臨床推奨曝露量[AUC0-24h]の約0.18%)で見られ.到着後の流産の増加.眼の融合または欠損(無眼症.小眼症).脊椎・あばら(あばらまたは骨化中心の追加.脊椎弓.あばらおよび胸骨).頭蓋骨(後頭部融合)および横隔膜(ヘルニア)が含まれていた。 外固定).重度の横隔膜の奇形(ヘルニア)などがあります。 その他.骨化不全や骨化欠如(椎骨/胸骨.肋骨.四肢).椎骨/胸骨.骨盤帯.肺.胸腺.肝臓.尿管.臍動脈などの異常や変種があります。 0.05mg/kg/dayの投与量では.上記の眼球.肋骨および尿管の異常の発生率は低いものでした。
発がん性: 関連する試験は実施されていない。
【ファーマコキネティクス】
オラパリブの剤形は.錠剤とカプセル剤(カプセル剤は中国では未申告)です。 錠剤の経口バイオアベイラビリティは.カプセルのそれよりも高いです。 母集団薬物動態解析の結果.300mg錠剤を1日2回投与した場合の定常状態の曝露量(AUC)は.400mgカプセルを1日2回投与した場合よりも77%高いことが示された。300mg錠剤単回投与後のオラパリブのAUCおよびCmaxの幾何平均値は42.0 μg*h/mL(n=204) および 5.8 μg/mL(n=204) であった。204)であり,300 mg錠を1日2回投与したときの定常状態のAUCおよびCmaxの幾何平均値はそれぞれ49.0 μg*h/mL(n = 227)および7.7 μg/mL(n = 227)であった。 オラパリブのPKは時間依存的であり.複数回の投与により定常状態のクリアランスが15%減少した。
吸収。
オラパリブの経口投与では.吸収が速く.血漿中のピーク濃度の中央値は通常投与後1.5時間で到達します。300mg錠を1日2回反復投与した場合.定常状態のAUC平均集積比は1.8となりました。
オラパリブの全身曝露量(単回投与AUC)は.用量範囲が25mgから450mgの場合.用量にほぼ比例して増加し.Cmaxは同じ用量範囲では用量増加にやや比例せず増加しました。
高脂肪食との併用により.オラパリブの吸収速度(tmaxが2.5時間遅延)が増加したが.オラパリブの吸収範囲に大きな変化はなかった(平均AUCが約8%増加)。
ディストリビューション。
オラパリブ300mg単回投与後のオラパリブの平均(±標準偏差)見かけの分布容積は158±136Lであり.in vitroでの蛋白結合率は約82%であった。
メタボリズム。
in vitro試験において.CYP3A4/5はオラパリブの代謝に主に関与する酵素であることが示されています。
女性患者では.プロトタイプのオラパリブを経口投与した後.血漿中に循環する放射能の大部分(70%)が14C-オラパリブで占められています。 14C-オラパリブは広範囲に代謝され.プロトタイプ薬剤は尿中で15%.糞便中で6%を占めています。 代謝のほとんどは酸化反応に起因し.生成された成分の多くは.その後グルコシノレートや硫酸塩の結合を受ける。
排他的。
オラパリブ300 mg単回投与時の平均血漿終末半減期(±標準偏差)は14.9 ± 8.2時間.見かけの血漿クリアランスは7.4 ± 3.9 L/hであった。
14C-olaparib単回投与後.与えられた放射能の86%が7日間の収集期間内に回収され.44%が尿から.42%が糞便から回収されました。 大半は代謝物として排泄された。
特殊な人口。
集団薬物動態解析において.患者の年齢.性別.体重.人種(白人.中国人.日本人を含む)は.有意な共変量ではありませんでした。
肝機能障害肝機能障害の試験では.軽度の肝機能障害患者(Child-Pugh分類A;n=9)にオラパリブを投与した場合.肝機能正常患者(n=13)と比較して.平均AUCが15%.平均Cmaxが13%増加しました。 軽度の肝障害はオラパリブのタンパク質結合に影響を及ぼさないため.血漿中の総露出量は遊離した薬剤を表しています。 中等度または重度の肝障害を有する患者におけるデータは入手できない。
腎臓障害。
腎機能障害に関する試験では.腎機能正常者(CLcr≧81mL/min;n=12)と比較して.オラパリブを服用した軽度腎機能障害者(Cockcroft-Gault式で定義.CLcr=51~80mL/min;n=13)は.平均AUC増加率が24%.平均Cmax0… sub>が15%増加し.中等度の腎障害患者(CLcr = 31-50 mL/min.n=13)ではオラパリブの服用によりAUCとCmaxの平均値がそれぞれ44%と26%増加しました。 オラパリブの血漿蛋白結合の程度とクレアチニンクリアランスの間に相関を示す証拠はなかった。 重度の腎障害または末期腎不全(CLcr≦30mL/min)の患者に関するデータはありません。
【ストレージ】。
30℃以下で保存してください。
【パッケージ】。
アルミニウム-アルミニウムブリスターパック.1箱56錠入り(7プレート)
アルミニウム-アルミニウムブリスターパック.1箱112錠入り(14枚入り)
[有効期限]。
36ヶ月
【エグゼクティブ・スタンダード】
輸入医薬品の登録に関する基準。
【承認番号】
会社名:AbbVie Deutschland GmbH & Co. KG
生産拠点住所:Knollstrasse 67061 Ludwigshafen, Germany
中国駐在員事務所住所:江蘇省無錫市新区黄山路2号
郵便番号:214028
品質に関する苦情:400 828 1755.800 828 1755
製品情報無償提供:400 820 8116, 800 820 8116
ファックス: 021-38723255
ウェブサイト:www.astrazeneca.com.cn.
[プロパティ]本製品はフィルムコーティングされた錠剤です
150mg:緑色から緑色/灰色.楕円形の両凸錠で.片面に「OP150」の刻印があり.もう片面は空白である。
100mg:黄色~濃黄色の楕円形の両凸錠で.片面に「OP100」の刻印があり.もう片面は空白である。
【効能・効果】本剤は.白金製剤を含む化学療法による完全寛解または部分寛解後の.白金製剤感受性の再発卵巣がん.卵管がん.原発性腹膜がんの成人患者の維持療法に適応されます。
【スペック】(1) 150 mg.(2) 100 mg
【用法・用量】。
本製品は.抗悪性腫瘍剤の使用に経験豊かな医師の監督のもとで使用する必要があります。
推奨される投与量。
本製品は.150mgと100mgのサイズがあります。
なお.減量する場合は.100mg錠を使用する。
患者は.白金製剤を含む化学療法終了後8週間以内に本剤の投与を開始し.病勢進行または許容できない毒性が発現するまで治療を継続する必要があります。
投与方法。
経口投与。 本製品は丸ごと飲み込み.錠剤は噛んだり.砕いたり.溶かしたり.割ったりしないでください。 本製品は.食事と一緒に.または空腹時に摂取することができます。
投与ミス。
患者が薬の服用を忘れた場合.次の服用は通常通り予定時刻に行うこと。
投与量調節。
有害事象について
吐き気.嘔吐.下痢.貧血などの有害事象に対処するため.治療の中断や投与量の減量を検討します。
減量が必要な場合は.250mg(150mg錠1錠.100mg錠1錠)を1日2回(1日総量500mgに相当)に減量する。
さらに減量が必要な場合は.200mg(100mg×2錠)を1日2回(1日総量400mgに相当)に減量して服用する。
チトクロムP450(CYP) em>)3Aインヒビター。
本剤を使用する場合.強または中程度のCYP3A阻害剤との併用は推奨されず.他の代替薬を検討する必要があります。 強力なCYP3A阻害剤との併用が必要な場合は.本剤の用量を1回100mg(100mg錠1錠)を1日2回(1日総量200mgに相当)に減量することが推奨される。 中間作用型CYP3A阻害剤の併用が必要な場合は.150mg(150mg錠1錠)を1日2回(1日総量300mgに相当)に減量することが望ましい(【注意事項】及び【薬物相互作用】を参照)。
特殊な集団に対する薬。
腎臓障害:本剤は軽度の腎障害(クレアチニンクリアランス51~80mL/min)の患者には用量を調節することなく使用できる。中等度の腎障害(クレアチニンクリアランス31~50mL/min)の患者には本剤200mg(100mg錠2錠)1日2回(1日総量400mg相当)が推奨用量となる;重度の腎障害の患者には使用不可である 重度の腎障害又は末期腎不全(クレアチニンクリアランス≦30mL/min)の患者における本剤の安全性及び有効性に関するデータはなく.使用は推奨されない([薬物動態]の項参照)。
肝機能障害:。
本剤は軽度の肝障害(Child-Pugh 分類 A)の患者でも用量を調節することなく使用できる([薬物動態]の項参照)。 中等度又は重度の肝障害患者における本剤の安全性及び有効性に関するデータはなく.本剤の使用は推奨されない(【薬物動態】の項参照)。
小児または青少年:。
小児および青年に対する本剤の安全性および有効性は確立しておらず.小児患者への投与は推奨されない。
高齢者(>65歳):。
高齢者では開始時の用量調節は必要ありません。 75歳以上の患者さんについては.限られた臨床データしかありません。
【副作用】。
ある医薬品の臨床試験で観察された副作用の発現率は.他の医薬品の臨床試験で観察された副作用の発現率と直接比較することはできませんし.臨床試験は様々な異なる条件の下で実施されるため.臨床現場で観察される発現率を反映するものではありません。
再発卵巣がんに対する維持療法。
卵巣がん患者558名(オラパリブ投与331名.プラセボ投与227名)を対象に実施した臨床試験において.以下の副作用が報告されています。
SOLO-2。
SOLO-2試験では.プラチナ感受性生殖細胞由来乳がん感受性変異(gBRCAm)卵巣がん患者における本剤の維持療法としての安全性が評価されました。 本試験はプラセボ対照二重盲検試験で.294名の患者さんに.疾患進行または忍容できない毒性が発現するまで.本剤300mg(150mg×2錠)を1日2回(n=195)またはプラセボ錠を1日2回(n=99)投与しています。 試験期間中央値は.本製品投与群で19.4カ月.プラセボ投与群で5.6カ月でした。 グレードを問わず副作用による治療中止は.プラセボ群の18%に対し.本製品投与群では45%に認められ.副作用による減量は.プラセボ群の3%に対し.本製品投与群では27%に認められました。 治療の中断や減量に至った主な副作用は.貧血(22%).好中球減少(9%).疲労・脱力感(8%)などでした。 投与中止に至った患者さんの割合は.プラセボ群2%に対し.本製品群11%でした。
表1は.SOLO-2試験において本製品を投与された患者の20%以上に発現した有害事象をまとめたものです。 表2に.本剤を投与されたSOLO-2投与患者のうち.少なくとも25%に発現した臨床検査値異常を示す。
表1 SOLO-2における副作用a(≥20%) strong>オラパリブ錠で治療された患者。
【スペック】(1) 150 mg.(2) 100 mg
【用法・用量】。
本製品は.抗悪性腫瘍剤の使用に経験豊かな医師の監督のもとで使用する必要があります。
推奨される投与量。
本製品は.150mgと100mgのサイズがあります。
なお.減量する場合は.100mg錠を使用する。
患者は.白金製剤を含む化学療法終了後8週間以内に本剤の投与を開始し.病勢進行または許容できない毒性が発現するまで治療を継続する必要があります。
投与方法。
経口投与。 本製品は丸ごと飲み込み.錠剤は噛んだり.砕いたり.溶かしたり.割ったりしないでください。 本製品は.食事と一緒に.または空腹時に摂取することができます。
投与ミス。
患者が薬の服用を忘れた場合.次の服用は通常通り予定時刻に行うこと。
投与量調節。
有害事象について
吐き気.嘔吐.下痢.貧血などの有害事象に対処するため.治療の中断や投与量の減量を検討します。
減量が必要な場合は.250mg(150mg錠1錠.100mg錠1錠)を1日2回(1日総量500mgに相当)に減量する。
さらに減量が必要な場合は.200mg(100mg×2錠)を1日2回(1日総量400mgに相当)に減量して服用する。
チトクロムP450(CYP) em>)3Aインヒビター。
本剤を使用する場合.強または中程度のCYP3A阻害剤との併用は推奨されず.他の代替薬を検討する必要があります。 強力なCYP3A阻害剤との併用が必要な場合は.本剤の用量を1回100mg(100mg錠1錠)を1日2回(1日総量200mgに相当)に減量することが推奨される。 中間作用型CYP3A阻害剤の併用が必要な場合は.150mg(150mg錠1錠)を1日2回(1日総量300mgに相当)に減量することが望ましい(【注意事項】及び【薬物相互作用】を参照)。
特殊な集団に対する薬。
腎臓障害:本剤は軽度の腎障害(クレアチニンクリアランス51~80mL/min)の患者には用量を調節することなく使用できる。中等度の腎障害(クレアチニンクリアランス31~50mL/min)の患者には本剤200mg(100mg錠2錠)1日2回(1日総量400mg相当)が推奨用量となる;重度の腎障害の患者には使用不可である 重度の腎障害又は末期腎不全(クレアチニンクリアランス≦30mL/min)の患者における本剤の安全性及び有効性に関するデータはなく.使用は推奨されない([薬物動態]の項参照)。
肝機能障害:。
本剤は軽度の肝障害(Child-Pugh 分類 A)の患者でも用量を調節することなく使用できる([薬物動態]の項参照)。 中等度又は重度の肝障害患者における本剤の安全性及び有効性に関するデータはなく.本剤の使用は推奨されない(【薬物動態】の項参照)。
小児または青少年:。
小児および青年に対する本剤の安全性および有効性は確立しておらず.小児患者への投与は推奨されない。
高齢者(>65歳):。
高齢者では開始時の用量調節は必要ありません。 75歳以上の患者さんについては.限られた臨床データしかありません。
【副作用】。
ある医薬品の臨床試験で観察された副作用の発現率は.他の医薬品の臨床試験で観察された副作用の発現率と直接比較することはできませんし.臨床試験は様々な異なる条件の下で実施されるため.臨床現場で観察される発現率を反映するものではありません。
再発卵巣がんに対する維持療法。
卵巣がん患者558名(オラパリブ投与331名.プラセボ投与227名)を対象に実施した臨床試験において.以下の副作用が報告されています。
SOLO-2。
SOLO-2試験では.プラチナ感受性生殖細胞由来乳がん感受性変異(gBRCAm)卵巣がん患者における本剤の維持療法としての安全性が評価されました。 本試験はプラセボ対照二重盲検試験で.294名の患者さんに.疾患進行または忍容できない毒性が発現するまで.本剤300mg(150mg×2錠)を1日2回(n=195)またはプラセボ錠を1日2回(n=99)投与しています。 試験期間中央値は.本製品投与群で19.4カ月.プラセボ投与群で5.6カ月でした。 グレードを問わず副作用による治療中止は.プラセボ群の18%に対し.本製品投与群では45%に認められ.副作用による減量は.プラセボ群の3%に対し.本製品投与群では27%に認められました。 治療の中断や減量に至った主な副作用は.貧血(22%).好中球減少(9%).疲労・脱力感(8%)などでした。 投与中止に至った患者さんの割合は.プラセボ群2%に対し.本製品群11%でした。
表1は.SOLO-2試験において本製品を投与された患者の20%以上に発現した有害事象をまとめたものです。 表2に.本剤を投与されたSOLO-2投与患者のうち.少なくとも25%に発現した臨床検査値異常を示す。
表1 SOLO-2における副作用a(≥20%) strong>オラパリブ錠で治療された患者。
| オラパリブ錠 | プラシーボ | |||
| 1-4 学年. % |
Grade 3-4Grade 3-4 Grade 3-4Grade 3-4Grade 3-4Grade 3-4 % |
第1学年から第4学年
. |
第3-4学年. % |
|
| 血液およびリンパ系の疾患 | ||||
| 貧血b | 44 | 20 | 9 | 2 |
| 消化器系の疾患 | ||||
| 吐き気 | 76 | 3 | 33 | 0 |
| 嘔吐 | 37 | 3 | 19 | 1 |
| 下痢 | 33 | 2 | 22 | 0 |
| 口腔粘膜炎c | ||||
| 口腔粘膜炎 | 20 | 1 | 16 | 0 |
| 感染症・伝染病 | ||||
| 上咽頭炎/上気道炎/副鼻腔炎/鼻炎/インフルエンザ | 上咽頭炎/上気道炎/鼻炎/インフルエンザ | 36 | 0 | 29 | 0 |
| 全身性疾患.投与部位の各種反応 | ||||
| 疲労感(脱力感を含む) | 66 | 4 | 39 | 2 |
| 代謝障害および栄養障害 | ||||
| 食欲不振 | 22 | 0 | 11 | 0 |
| 様々な筋骨格系および結合組織障害 | ||||
| 関節痛/筋肉痛 | 30 | 0 | 28 | 0 |
| あらゆる種類の神経障害 | ||||
| 味覚障害 | 27 | 0 | 7 | 0 |
| 頭痛 | 26 | 1 | 14 | 0 |
。
a米国国立がん研究所有害事象評価基準(CTCAE)バージョン4.0に基づく評定。
bは.貧血.赤血球圧容量減少.ヘモグロビン減少.鉄欠乏症.平均細胞量増加.赤血球数減少などのカテゴリー項を表しています。
c は.口腔膿瘍.口内炎.歯肉膿瘍.歯肉疾患.歯肉痛.口腔潰瘍.粘膜感染.粘膜炎症.口腔カンジダ症.口腔不快感.口腔ヘルペス.口腔感染.口腔粘膜紅斑.口腔痛.中咽頭不快感.中咽頭痛などのカテゴリー用語のことです。
また.SOLO-2試験において本剤投与患者の20%に発現した副作用は.好中球減少.発疹.咳.消化不良.白血球減少.低マグネシウム血症.めまい.血小板減少.血清クレアチニン上昇.リンパ球減少及び浮腫であった。
表2 SOLO-2患者の25%以上で報告された臨床検査値異常
…….
| Laboratory indicatorsa | オラパリブ錠 | Placebo nb=99 |
||
| 1-4 学年。 % |
第3-4学年
. |
第1学年から第4学年
. |
第3-4学年. % |
|
| 平均赤血球容積の増加c | 89 | – | 52 | – |
| ヘモグロビンの低下 | 83 | 17 | 69 | 0 |
| 白血球数の低下 | 69 | 5 | 48 | 1 |
| リンパ球数の減少 | 67 | 11 | 37 | 1 |
| 絶対好中球数の減少 | 51 | 7 | 34 | 3 |
| 血清クレアチニン値上昇 | 44 | 0 | 29 | 0 |
| 血小板数の低下 | 42 | 2 | 22 | 1 |
。
a 臨床検査値がCTCAE grade 1の患者は.臨床試験への登録が許可されます。
b 安全性データセットの人数を表し.表中のデータは評価可能なすべての患者さんの各検査項目に基づいています。
c は.平均赤血球容積が正常上限値(ULN)である被験者の割合を表す。
勉強19。
オラパリブカプセルが投与された患者のうち.副作用による治療中断は35%(プラセボ群10%).減量が必要な患者は26%(同4%).中止となった患者は6%(同2%)であり.プラセボ群に比べ.副作用は少なかった。
表3は.19試験においてオラパリブ投与患者の20%以上に発現した有害事象をまとめたものです。 表4は.19試験でオラパリブを投与された患者の25%以上に発生した臨床検査値異常の一覧です。
表3 試験における副作用 19a 11
. strong>(オラパリブを投与された患者の20%以上)。
| 副作用 | オラパリブカプセル. n=136 |
プラシーボ | ||
| 1-4 学年. % |
Grade 3-4Grade 3-4 Grade 3-4Grade 3-4Grade 3-4Grade 3-4 % |
第1学年から第4学年
. |
第3-4学年. % |
|
| 血液およびリンパ系の疾患 | ||||
| 貧血b | 23 | 7 | 7 | 1 |
| 消化器系の疾患 | ||||
| 吐き気 | 71 | 2 | 36 | 0 |
| 嘔吐 | 35 | 2 | 14 | 1 |
| 下痢 | 28 | 2 | 25 | 2 |
| 便秘 | 22 | 1 | 12 | 0 |
| 全身性疾患.投与部位の各種反応 | ||||
| 疲労感(脱力感を含む) | 63 | 9 | 46 | 3 |
| 感染症・伝染病 | ||||
| 呼吸器感染症 | 22 | 2 | 11 | 0 |
| 代謝障害および栄養障害 | 代謝障害および栄養障害 | |||
| 食欲不振 | 21 | 0 | 13 | 0 |
| あらゆる種類の神経障害 | ||||
| 頭痛 | 21 | 0 | 13 | 1 |
a 米国国立がん研究所(NCI)CTCAE 4.0分類による。
b 医学的な副作用の概念を反映した関連用語を表すカテゴリー用語。
また.19試験で本剤が投与された患者のうち20%に.消化不良.口腔粘膜炎.味覚異常.めまい.血中クレアチニン上昇.好中球減少.血小板減少.白血球減少.リンパ球減少.呼吸困難.発熱.浮腫等の副作用が認められました。
表4 試験参加者の25%以上が報告した検査項目 19 1. 検査異常。
| Laboratory indicatorsa | オラパリブカプセル | プラシーボ | ||
| 第1学年から第4学年
. |
第3-4学年
. |
第1学年から第4学年。 % |
第3-4学年. % |
|
| ヘモグロビンの減少 | 82 | 8インチ | 58 | 1 |
| 平均赤血球容積の増加c | 82 | – | 51 | – |
| 白血球数の低下 | 58 | 4 | 37 | 2 |
| リンパ球数の減少 | 52 | 10 | 32 | 3 |
| 絶対好中球数の減少 | 47 | 7 | 40 | 2 |
| 血清クレアチニン値上昇 | 45 | 0 | 14 | 0 |
| 血小板数の低下 | 36 | 4 | 18 | 0 |
。
a 臨床検査値がCTCAE grade 1の患者は.臨床試験への登録が許可されます。
b 安全性データセットの人数を表し.表中のデータは評価可能なすべての患者さんの各検査項目に基づいています。
c は.平均赤血球容積が正常上限値(ULN)である被験者の割合を表す。
特定の副作用の説明。
血液学的毒性。
貧血やその他の血液毒性は全体的に低グレード(CTCAE グレード 1 または 2)でしたが.CTCAE グレード 3 以上の事象も報告されています。
貧血が初めて発現するまでの期間の中央値は約4週間(CTCAE≧グレード3の事象は約7週間)です。 貧血は.治療の中断および投与量の減量([用法・用量]を参照).および適切な場合には輸血によってコントロールすることができます。 SOLO2試験において.貧血の発生率は43.6%(CTCAE≧3では19.5%).貧血による投与中断.減量.中止の発生率はそれぞれ16.9%.8.2%.3.1%で.オラパリブ投与患者の17.9%が1回以上の輸血を必要としています。 オラパリブとヘモグロビン減少の間に量的効果関係が証明されています。 オラパリブ錠の臨床試験において.ベースラインに対するヘモグロビンの変化(低下)CTCAEグレード≧2の発生率は20%.好中球の絶対値は15%.血小板は5%.リンパ球は30%.白血球は20%(%はいずれも概数%)となっています。
平均赤血球容積がベースライン時の低値または正常値から正常上限値以上に増加した発現率は約55%であり.投与中止後に正常値に戻り.臨床的に重大な影響を及ぼすことはなかった。
治療開始後12ヶ月間は.ベースライン時の全血球数検査とその後の毎月のモニタリングが推奨され.その後は治療中に生じた臨床的に意味のあるパラメータの変化について定期的にモニタリングする。 これらの変化により.投与の中断または減量.さらなる治療が必要となる場合がある([用法]および[使用上の注意]を参照)。
その他の検査結果。
本製品の臨床試験において.血中クレアチニン値のベースラインからのCTCAE≧Grade 2の上昇の発生率は約15%でした。 プラセボ対照二重盲検比較試験のデータでは.血中クレアチニン値の中央値は投与前と比較して23%上昇し.投与中は維持され.投与中止後はベースラインに戻り.臨床的に重大な後遺症は認められませんでした。
吐き気と嘔吐。
吐き気は通常.治療のごく初期に発生し.ほとんどの患者さんで治療開始後1ヵ月以内に最初の吐き気が起こります。 嘔吐は通常.治療開始後早期に発生し.大多数の患者さんで治療開始後2ヶ月以内に最初の嘔吐が起こります。 吐き気および嘔吐は.大多数の患者で断続的であると報告されており.投与の中断.投与量の削減および/または制吐療法により制御することができます。 予防的制吐剤は必要ありません。
【禁忌】本剤の有効成分または賦形剤成分に対して過敏症のある人は禁忌である。 治療中および最終投与後1カ月間は授乳を中止させること([妊婦・授乳婦等への使用]の項参照)。
【注意事項】血液学的毒性。
本剤を投与された患者において.軽度または中等度(CTCAEグレード1または2)の貧血.好中球減少.血小板減少およびリンパ球減少の臨床診断および/または検査所見を含む血液学的毒性が報告されています。 患者は.以前の抗悪性腫瘍剤治療による血液学的毒性が回復するまで本剤の投与を開始しないこと(ヘモグロビン.血小板及び好中球のレベルがCTCAEグレード1以下まで回復すること)。 治療開始後12ヶ月間はベースライン時に全血球検査を行い.その後毎月モニタリングを行い.治療中に生じた臨床的に重要なパラメータの変化については.その後も定期的にモニタリングを行うことが推奨されます([副作用]を参照)。
重篤な血液毒性または輸血依存性の血液毒性が発現した場合は.治療を中断し.関連する血液学的検査を実施すること。 本剤の投与を4週間中断しても臨床的に異常な血液学的パラメータが残存する場合.骨髄検査及び/又は血液細胞遺伝学的検査を行うことが推奨される。
骨髄異形成症候群/急性骨髄性白血病と呼ばれるもの。
長期生存期間の追跡調査を含む臨床試験において.本剤単独投与患者における骨髄異形成症候群/急性骨髄性白血病(MDS/AML)の発現率は.<1.5%でした。 イベントの大半は.死亡という結果をもたらしました。 MDS/AMLを発症した患者において.オラパリブの投与期間は6ヶ月未満から2年以上と幅があり.より長い曝露期間に関するデータは限られています。 すべての患者はMDS/AMLの基礎因子を有し.白金製剤ベースの化学療法の前治療を受けていた。 一部の患者は.放射線治療だけでなく.DNAに損傷を与える他の薬物による治療も受けていた。 患者の大半はgBRCA 1/2変異のキャリアであった。 これらの患者さんの中には.過去に腫瘍や骨髄異形成の病歴がある方もいらっしゃいます。 オラパリブ錠の治療中にMDSおよび/またはAMLと診断された場合.オラパリブ錠の治療を中止し.適切な治療を行うことが推奨されます。
非感染性肺炎。
本剤単独投与時の臨床試験における非感染性肺炎(死亡に至る事象を含む)の発現率は1.0%でした。 報告された非感染性肺炎の臨床症状は様々で.多くの病因(肺がんおよび/または転移性肺がん.基礎疾患である肺疾患.喫煙歴および/または化学療法や放射線療法の既往歴)に影響されていた。 呼吸困難.咳嗽.発熱等の呼吸器症状が新たに発現又は悪化した場合.又は胸部画像所見が異常な場合は.治療を一時中断し.直ちに関連する検査を開始すること。 非感染性肺炎と診断された場合は.治療を中止し.適切な処置を行うこと。
胚性–胎児への毒性。
本剤の作用機序(ポリADPリボースポリメラーゼ阻害作用)及び動物実験から.妊婦に投与した場合.胎児に害を与える可能性がある。 ラットを用いた前臨床試験において.オラパリブの胚・胎児生存率への悪影響が示されています。 ヒトの推奨用量である1日2回300mg以下の曝露では.重度の胎児奇形が誘発される可能性があります。
本製品は.妊娠中は服用しないでください。 本剤服用中に患者が妊娠した場合には.胎児への危険性があることを患者に説明する必要がある。 妊娠可能な年齢の女性は.治療中および最終投与後6ヶ月間は効果的な避妊をすることが推奨されます。
男性患者及び妊娠可能な年齢の女性パートナーには.治療中及び最終投与後3カ月間は効果的な避妊を行い.精子提供を行わないよう助言すること([妊娠中及び授乳中の女性における使用]の項参照)。
他の医薬品との相互作用。
本剤と強又は中等度のCYP3A阻害剤との併用は推奨されない([用法・用量]を参照)。 強力または中程度のCYP3A阻害剤との併用が必要な場合は.投与量を減らす必要がある([用法・用量]を参照)。
本剤と強力または中間作用型の CYP3A 誘導剤との併用は推奨されない。 本剤の投与を既に受けている患者で.強力又は中間作用性CYP3A誘導剤による治療を必要とする場合.本剤の有効性が著しく低下する可能性があることを処方医は認識すべきである([薬物相互作用]の項参照)。
運転や機械操作の能力への影響。
オラパリブの運転や機械操作の能力への影響に関する研究は行われていません。 しかし.本剤投与中に脱力感.疲労感.めまいが報告されており.これらの症状が現れた患者さんは.運転や機械の操作に注意してください。
効果QTインターバル。
オラパリブの心臓再分極への影響は.300mg単回投与後の119名と300mg1日2回複数回投与後の109名で評価されました。 オラパリブのQT間隔に対する臨床的な関連性は認められませんでした。
[妊娠・授乳期について
【妊娠中・授乳中の方の使用】。
避妊。
妊娠可能な年齢の女性は.本製品による治療の開始時および治療中に妊娠してはならず.治療を開始する前に妊娠検査が必要です。 妊娠可能な女性は.治療中および本製品の最終投与後6カ月間は有効な避妊を行う必要があります([使用上の注意]を参照)。 オラパリブは酵素誘導によりCYP2C9基質への曝露を減少させる可能性があり.オラパリブとの併用によりホルモン避妊薬の効果が減少する可能性を否定できません。 したがって.治療中は他の非ホルモン性避妊手段を考慮し.定期的に妊娠検査を行う必要があります([薬物相互作用]の項参照)。
本剤の遺伝毒性試験及び動物生殖毒性試験に基づき.男性患者(配偶者が妊娠可能な年齢の女性又は妊婦)は.治療中及びオラパリブ最終投与後3カ月間は有効な避妊を行い.精子提供を行わないことが推奨されている(【注意事項】を参照)。
妊娠。
動物実験では.母体の全身曝露量が治療用量におけるヒトの曝露量よりも低いラット試験において.重度の催奇形性作用及び胚・胎児生存率への影響を含む生殖毒性が示されている([薬理学及び毒性]を参照)。 妊婦へのオラパリブの使用に関するデータはありませんが.オラパリブの作用機序に基づき.妊娠可能な年齢で確実な避妊を行っていない女性には.治療中および本製品の最終投与後6カ月間はオラパリブ錠を使用しないでください。
授乳。
オラパリブの母乳中への分泌に関する動物実験は実施されていない。 オラパリブ又はその代謝物がヒト母乳中に分泌されるかどうかは不明である。 本剤の薬理学的プロファイルに基づき.オラパリブ錠剤による治療中及び最終投与後1カ月間は授乳を中止することが望ましい([禁忌]を参照)。
妊産婦。
生殖能力に関する臨床データは得られていない。 動物実験では.被験薬は妊娠に影響を与えないが.胚・胎児の生存に悪影響を及ぼすことが示されている(【薬理作用と毒性】参照)。
【注意事項】血液学的毒性。
本剤を投与された患者において.軽度または中等度(CTCAEグレード1または2)の貧血.好中球減少.血小板減少およびリンパ球減少の臨床診断および/または検査所見を含む血液学的毒性が報告されています。 患者は.以前の抗悪性腫瘍剤治療による血液学的毒性が回復するまで本剤の投与を開始しないこと(ヘモグロビン.血小板及び好中球のレベルがCTCAEグレード1以下まで回復すること)。 治療開始後12ヶ月間はベースライン時に全血球検査を行い.その後毎月モニタリングを行い.治療中に生じた臨床的に重要なパラメータの変化については.その後も定期的にモニタリングを行うことが推奨されます([副作用]を参照)。
重篤な血液毒性または輸血依存性の血液毒性が発現した場合は.治療を中断し.関連する血液学的検査を実施すること。 本剤の投与を4週間中断しても臨床的に異常な血液学的パラメータが残存する場合.骨髄検査及び/又は血液細胞遺伝学的検査を行うことが推奨される。
骨髄異形成症候群/急性骨髄性白血病と呼ばれるもの。
長期生存期間の追跡調査を含む臨床試験において.本剤単独投与患者における骨髄異形成症候群/急性骨髄性白血病(MDS/AML)の発現率は.<1.5%でした。 イベントの大半は.死亡という結果をもたらしました。 MDS/AMLを発症した患者において.オラパリブの投与期間は6ヶ月未満から2年以上と幅があり.より長い曝露期間に関するデータは限られています。 すべての患者はMDS/AMLの基礎因子を有し.白金製剤ベースの化学療法の前治療を受けていた。 一部の患者は.放射線治療だけでなく.DNAに損傷を与える他の薬物による治療も受けていた。 患者の大半はgBRCA 1/2変異のキャリアであった。 これらの患者さんの中には.過去に腫瘍や骨髄異形成の病歴がある方もいらっしゃいます。 オラパリブ錠の治療中にMDSおよび/またはAMLと診断された場合.オラパリブ錠の治療を中止し.適切な治療を行うことが推奨されます。
非感染性肺炎。
本剤単独投与時の臨床試験における非感染性肺炎(死亡に至る事象を含む)の発現率は1.0%でした。 報告された非感染性肺炎の臨床症状は様々で.多くの病因(肺がんおよび/または転移性肺がん.基礎疾患である肺疾患.喫煙歴および/または化学療法や放射線療法の既往歴)に影響されていた。 呼吸困難.咳嗽.発熱等の呼吸器症状が新たに発現又は悪化した場合.又は胸部画像所見が異常な場合は.治療を一時中断し.直ちに関連する検査を開始すること。 非感染性肺炎と診断された場合は.治療を中止し.適切な処置を行うこと。
胚性–胎児への毒性。
本剤の作用機序(ポリADPリボースポリメラーゼ阻害作用)及び動物実験から.妊婦に投与した場合.胎児に害を与える可能性がある。 ラットを用いた前臨床試験において.オラパリブの胚・胎児生存率への悪影響が示されています。 ヒトの推奨用量である1日2回300mg以下の曝露では.重度の胎児奇形が誘発される可能性があります。
本製品は.妊娠中は服用しないでください。 本剤服用中に患者が妊娠した場合には.胎児への危険性があることを患者に説明する必要がある。 妊娠可能な年齢の女性は.治療中および最終投与後6ヶ月間は効果的な避妊をすることが推奨されます。
男性患者及び妊娠可能な年齢の女性パートナーには.治療中及び最終投与後3カ月間は効果的な避妊を行い.精子提供を行わないよう助言すること([妊娠中及び授乳中の女性における使用]の項参照)。
他の医薬品との相互作用。
本剤と強又は中等度のCYP3A阻害剤との併用は推奨されない([用法・用量]を参照)。 強力または中程度のCYP3A阻害剤との併用が必要な場合は.投与量を減らす必要がある([用法・用量]を参照)。
本剤と強力または中間作用型の CYP3A 誘導剤との併用は推奨されない。 本剤の投与を既に受けている患者で.強力又は中間作用性CYP3A誘導剤による治療を必要とする場合.本剤の有効性が著しく低下する可能性があることを処方医は認識すべきである([薬物相互作用]の項参照)。
運転や機械操作の能力への影響。
オラパリブの運転や機械操作の能力への影響に関する研究は行われていません。 しかし.本剤投与中に脱力感.疲労感.めまいが報告されており.これらの症状が現れた患者さんは.運転や機械の操作に注意してください。
効果QTインターバル。
オラパリブの心臓再分極への影響は.300mg単回投与後の119名と300mg1日2回複数回投与後の109名で評価されました。 オラパリブのQT間隔に対する臨床的な関連性は認められませんでした。
[妊娠・授乳期について
【妊娠中・授乳中の方の使用】。
避妊。
妊娠可能な年齢の女性は.本製品による治療の開始時および治療中に妊娠してはならず.治療を開始する前に妊娠検査が必要です。 妊娠可能な女性は.治療中および本製品の最終投与後6カ月間は有効な避妊を行う必要があります([使用上の注意]を参照)。 オラパリブは酵素誘導によりCYP2C9基質への曝露を減少させる可能性があり.オラパリブとの併用によりホルモン避妊薬の効果が減少する可能性を否定できません。 したがって.治療中は他の非ホルモン性避妊手段を考慮し.定期的に妊娠検査を行う必要があります([薬物相互作用]の項参照)。
本剤の遺伝毒性試験及び動物生殖毒性試験に基づき.男性患者(配偶者が妊娠可能な年齢の女性又は妊婦)は.治療中及びオラパリブ最終投与後3カ月間は有効な避妊を行い.精子提供を行わないことが推奨されている(【注意事項】を参照)。
妊娠。
動物実験では.母体の全身曝露量が治療用量におけるヒトの曝露量よりも低いラット試験において.重度の催奇形性作用及び胚・胎児生存率への影響を含む生殖毒性が示されている([薬理学及び毒性]を参照)。 妊婦へのオラパリブの使用に関するデータはありませんが.オラパリブの作用機序に基づき.妊娠可能な年齢で確実な避妊を行っていない女性には.治療中および本製品の最終投与後6カ月間はオラパリブ錠を使用しないでください。
授乳。
オラパリブの母乳中への分泌に関する動物実験は実施されていない。 オラパリブ又はその代謝物がヒト母乳中に分泌されるかどうかは不明である。 本剤の薬理学的プロファイルに基づき.オラパリブ錠剤による治療中及び最終投与後1カ月間は授乳を中止することが望ましい([禁忌]を参照)。
妊産婦。
生殖能力に関する臨床データは得られていない。 動物実験では.被験薬は妊娠に影響を与えないが.胚・胎児の生存に悪影響を及ぼすことが示されている(【薬理作用と毒性】参照)。
【小児用】小児および青年に対する本剤の安全性および有効性は確立していないため.本剤は小児用としては不適当である。
【老人用】オラパリブ錠300mg1日2回単剤投与を受けた進行性固形癌患者687例を対象とした臨床試験において.146例(21%)が65歳以上.うち29例(4%)が75歳以上.85歳以上の患者さんはいませんでした。 オラパリブによる治療の安全性と有効性のデータには.若年層と高齢層の間で有意な差は見られませんでした。
[薬物相互作用
薬力学的相互作用。
本剤とDNAに損傷を与える薬剤を含む他の抗悪性腫瘍剤との併用による臨床試験では.骨髄抑制毒性のレベルの上昇と持続時間の延長が確認されています。 骨髄抑制作用を有する抗悪性腫瘍剤との併用には.単剤での推奨用量は適用されません。
オラパリブとワクチンや免疫抑制剤との併用試験は実施されていません。 したがって.上記の薬剤とオラパリブ錠の併用には注意が必要であり.患者の状態を十分に観察する必要があります。
ファーマコキネティック相互作用。
In vitro試験において.オラパリブはCYP3Aの阻害剤およびCYP2B6の誘導剤であることが確認されています。 オラパリブは.ヒトでは弱いCYP3A阻害剤であると予測されます。 In vitroの研究では.オラパリブが.ウリジン二リン酸グルクロニルトランスフェラーゼ(UGT)1A1.乳癌耐性タンパク質(BCRP).有機アニオン輸送タンパク質(OATP)1B1.有機カチオン輸送タンパク質(OCT)1.OCT2.有機アニオン輸送タンパク質(OAT)3.多剤・毒物排出輸送タンパク質(MATE)1およびMATE2Kの阻害剤であるかも示された。 の阻害剤である。 これらの知見の臨床的な関連性は不明である。 In vitro 試験において.オラパリブは排出トランスポーターである P-glycoprotein (P-gp) の基質であり.P-gp を阻害する。 オラパリブが P-gp を誘導する可能性は評価されていない。
オラパリブの血漿中濃度を上昇させる可能性がある薬剤。
オラパリブは.主にCYP3Aによって代謝されます。 患者(n=57)において.強力なCYP3A阻害剤であるイトラコナゾールの併用により.オラパリブのAUCが170%増加しました。 中活性型CYP3A阻害剤フルコナゾールは.オラパリブの血漿中濃度対時間曲線下面積(AUC)を121%増加させると予測された。
イトラコナゾール.テリスロマイシン.クラリスロマイシン.ケトコナゾール.ボリコナゾール.ネファゾドン.ポサコナゾール.リトナビル.ロピナビル/リトナビル.インジナビル.サキナビル.ネルフィナビル.ボセプレビル.テラプレビル等の強力なCYP3A阻害剤やアンプレナビル等のCYPA中間阻害剤の併用は避け.また.これらの阻害剤と併用することで.より高い効果が期待できます。 (アンプレナビル).アリピタント.アタザナビル.シプロフロキサシン.クリゾチニブ.ダルナビル/リトナビル.ジルチアゼム.エリスロマイシン.フルコナゾール.ホスアンプレナビル.イマチニブ.ベラパミル。 強または中程度のCYP3A阻害剤の併用が必要な場合は.オラパリブの投与量を減量する必要があります。
グレープフルーツ.グレープフルーツジュース.ライム.ライムジュースはCYP3A阻害剤を含むため.オラパリブ治療中は避けてください。
オラパリブの血漿中濃度を低下させる可能性のある薬剤。
患者(n=22)では.強力なCYP3A誘導物質であるリファンピシンの併用により.オラパリブのAUCが87%減少しました。 中活性型CYP3A誘導剤は.オラパリブのAUCを約60%減少させると予測された。
フェニトイン.リファンピシン.カルバマゼピン.セントジョーンズワートなどの強力なCYP3A誘導剤).ボセンタン.エファビレンツ.エトラビリン.モダフィニル.ナフシリンなどの中間作用型CYP3A4誘導剤の併用は避けてください。 中間作用型CYP3A誘導剤の使用を回避できない場合.オラパリブの有効性が低下する可能性があります。
[薬物相互作用
薬力学的相互作用。
本剤とDNAに損傷を与える薬剤を含む他の抗悪性腫瘍剤との併用による臨床試験では.骨髄抑制毒性のレベルの上昇と持続時間の延長が確認されています。 骨髄抑制作用を有する抗悪性腫瘍剤との併用には.単剤での推奨用量は適用されません。
オラパリブとワクチンや免疫抑制剤との併用試験は実施されていません。 したがって.上記の薬剤とオラパリブ錠の併用には注意が必要であり.患者の状態を十分に観察する必要があります。
ファーマコキネティック相互作用。
In vitro試験において.オラパリブはCYP3Aの阻害剤およびCYP2B6の誘導剤であることが確認されています。 オラパリブは.ヒトでは弱いCYP3A阻害剤であると予測されます。 In vitroの研究では.オラパリブが.ウリジン二リン酸グルクロニルトランスフェラーゼ(UGT)1A1.乳癌耐性タンパク質(BCRP).有機アニオン輸送タンパク質(OATP)1B1.有機カチオン輸送タンパク質(OCT)1.OCT2.有機アニオン輸送タンパク質(OAT)3.多剤・毒物排出輸送タンパク質(MATE)1およびMATE2Kの阻害剤であるかも示された。 の阻害剤である。 これらの知見の臨床的な関連性は不明である。 In vitro 試験において.オラパリブは排出トランスポーターである P-glycoprotein (P-gp) の基質であり.P-gp を阻害する。 オラパリブが P-gp を誘導する可能性は評価されていない。
オラパリブの血漿中濃度を上昇させる可能性がある薬剤。
オラパリブは.主にCYP3Aによって代謝されます。 患者(n=57)において.強力なCYP3A阻害剤であるイトラコナゾールの併用により.オラパリブのAUCが170%増加しました。 中活性型CYP3A阻害剤フルコナゾールは.オラパリブの血漿中濃度対時間曲線下面積(AUC)を121%増加させると予測された。
イトラコナゾール.テリスロマイシン.クラリスロマイシン.ケトコナゾール.ボリコナゾール.ネファゾドン.ポサコナゾール.リトナビル.ロピナビル/リトナビル.インジナビル.サキナビル.ネルフィナビル.ボセプレビル.テラプレビル等の強力なCYP3A阻害剤やアンプレナビル等のCYPA中間阻害剤の併用は避け.また.これらの阻害剤と併用することで.より高い効果が期待できます。 (アンプレナビル).アリピタント.アタザナビル.シプロフロキサシン.クリゾチニブ.ダルナビル/リトナビル.ジルチアゼム.エリスロマイシン.フルコナゾール.ホスアンプレナビル.イマチニブ.ベラパミル。 強または中程度のCYP3A阻害剤の併用が必要な場合は.オラパリブの投与量を減量する必要があります。
グレープフルーツ.グレープフルーツジュース.ライム.ライムジュースはCYP3A阻害剤を含むため.オラパリブ治療中は避けてください。
オラパリブの血漿中濃度を低下させる可能性のある薬剤。
患者(n=22)では.強力なCYP3A誘導物質であるリファンピシンの併用により.オラパリブのAUCが87%減少しました。 中活性型CYP3A誘導剤は.オラパリブのAUCを約60%減少させると予測された。
フェニトイン.リファンピシン.カルバマゼピン.セントジョーンズワートなどの強力なCYP3A誘導剤).ボセンタン.エファビレンツ.エトラビリン.モダフィニル.ナフシリンなどの中間作用型CYP3A4誘導剤の併用は避けてください。 中間作用型CYP3A誘導剤の使用を回避できない場合.オラパリブの有効性が低下する可能性があります。
【薬物過剰摂取】本製品による過量投与の臨床的徴候は確認されていない。 オラパリブ錠を1日900mgまで2日間服用した少数の患者において.予期せぬ副作用は報告されていません。 過量投与に対する特別な管理はありません。 過量投与が発生した場合.医師は対症療法および一般的な支持療法を行う必要があります。
【臨床試験】再発卵巣癌に対する維持療法。
プラチナ製剤を含む治療で寛解を得た再発卵巣癌患者を対象とした2つの無作為化プラセボ対照二重盲検多施設共同試験で.olaparibの有効性が評価されました。
SOLO2研究 (D0816C00002) 研究 () 研究 () 研究 ()
SOLO2試験は.gBRCA1/2変異を有するプラチナ感受性再発卵巣がん.卵管がん.原発性腹膜がんの患者さんにおける維持療法としてのオラパリブの安全性と有効性を評価した無作為化二重盲検プラセボ対照第III相試験です。 白金製剤を含む化学療法終了後に寛解(完全寛解[CR]または部分寛解[PR])した高悪性度漿液性または内膜性PSR卵巣がん患者295名を登録し.2対1の割合で(オラパリブ群196名.プラセボ群99名).病勢進行または以下の症状が発現するまでオラパリブ(300mg[150mg錠2]1日2回)またはプラセボの投与を行いました。 耐容性のない毒性
2種類以上の白金製剤を含む化学療法を受けた患者は.最後の白金製剤を含む化学療法終了後6ヶ月以内に疾患が再発するまで研究に参加しました。 オラパリブまたは他のPARP阻害剤による前治療を受けていない患者であること。 ベバシズマブによる前治療歴は認められますが.無作為化前の治療レジメンにベバシズマブが含まれていないことが条件となります。
ベースライン時.すべての患者が.局所検査(n=236)または中央Myriad CLIA(n=59)で.その後BRACAnalysis® CDx(n=286)で確認したgBRCA 変異を有しているか.または変異が疑われる患者。 無作為化された患者の4.7%(14/295人)でBRCA1/2遺伝子にラージフラグメント再配列が検出された。
人口統計学的特性およびベースラインの特性は.オラパリブ群とプラセボ群でほぼ同じでした。 両群の年齢の中央値は56歳であった。 卵巣がんが80%以上の患者さんの原発であった。 最も多い組織型はplasmacytic (> 90%)で.6%の患者にはendometrioid carcinomaが見られた。 オラパリブ群では.過去に二次治療のみを受けていた患者さんが55%.三次治療以上を受けていた患者さんが45%でした。 プラセボ群では.61%の患者さんが二次治療のみ.39%の患者さんが三次治療以上の治療を受けていました。 白金製剤を含む化学療法への反応性は.完全寛解が47%.部分寛解が53%であり.ECOG fitness scoreは0が81%と大半の症例で認められた。 オラパリブ群とプラセボ群では.それぞれ17%と20%の患者さんがベバシズマブによる前治療を受けていました。
主要評価項目は無増悪生存期間(PFS)で.RECIST(Remission Evaluation Criteria in Solid Tumours)1.1評価法を用いて治験医師により測定されました。 副次的評価項目は.二次疾患進行または死亡までの期間(PFS2).OS(全生存期間).治療中止または死亡までの期間(TDT).最初の抗がん剤治療開始または死亡までの期間(TFST).二次抗がん剤治療開始または死亡までの期間(TSST).HRQoL(健康に関する生活の質)とし.これらの評価項目が達成されたことを確認した上で.本試験の結果を発表しました。 ).
本試験では.主要評価項目であるPFSがプラセボ群に対してオラパリブ群で統計的に有意に改善し.リスク比(HR)は0.30(95%CI 0.22-0.41; p<0.0001; 中央値はオラパリブ群19.1カ月に対してプラセボ群5.5カ月)であった。 治験責任医師が評価したPFSの結果は.盲検化された独立した中央画像評価によって裏付けられました(HR=0.25.95% CI 0.18-0.35.p<=0.0001.中央値はオラパリブ群30.2カ月.プラセボ群5.5カ月)。2年後.プラセボ群と比較してオラパリブ投与群の43%が無進行を維持しました。 は15%にとどまりました。
SOLO2におけるgBRCA1/2m PSR卵巣がん患者様の主要評価項目の結果概要については.表5および図1をご覧ください。
表5 SOLO2におけるgBRCA1/2m PSR 卵巣癌患者の主要評価項目。 試験結果の概要(治験責任医師による評価)。
【臨床試験】再発卵巣癌に対する維持療法。
プラチナ製剤を含む治療で寛解を得た再発卵巣癌患者を対象とした2つの無作為化プラセボ対照二重盲検多施設共同試験で.olaparibの有効性が評価されました。
SOLO2研究 (D0816C00002) 研究 () 研究 () 研究 ()
SOLO2試験は.gBRCA1/2変異を有するプラチナ感受性再発卵巣がん.卵管がん.原発性腹膜がんの患者さんにおける維持療法としてのオラパリブの安全性と有効性を評価した無作為化二重盲検プラセボ対照第III相試験です。 白金製剤を含む化学療法終了後に寛解(完全寛解[CR]または部分寛解[PR])した高悪性度漿液性または内膜性PSR卵巣がん患者295名を登録し.2対1の割合で(オラパリブ群196名.プラセボ群99名).病勢進行または以下の症状が発現するまでオラパリブ(300mg[150mg錠2]1日2回)またはプラセボの投与を行いました。 耐容性のない毒性
2種類以上の白金製剤を含む化学療法を受けた患者は.最後の白金製剤を含む化学療法終了後6ヶ月以内に疾患が再発するまで研究に参加しました。 オラパリブまたは他のPARP阻害剤による前治療を受けていない患者であること。 ベバシズマブによる前治療歴は認められますが.無作為化前の治療レジメンにベバシズマブが含まれていないことが条件となります。
ベースライン時.すべての患者が.局所検査(n=236)または中央Myriad CLIA(n=59)で.その後BRACAnalysis® CDx(n=286)で確認したgBRCA 変異を有しているか.または変異が疑われる患者。 無作為化された患者の4.7%(14/295人)でBRCA1/2遺伝子にラージフラグメント再配列が検出された。
人口統計学的特性およびベースラインの特性は.オラパリブ群とプラセボ群でほぼ同じでした。 両群の年齢の中央値は56歳であった。 卵巣がんが80%以上の患者さんの原発であった。 最も多い組織型はplasmacytic (> 90%)で.6%の患者にはendometrioid carcinomaが見られた。 オラパリブ群では.過去に二次治療のみを受けていた患者さんが55%.三次治療以上を受けていた患者さんが45%でした。 プラセボ群では.61%の患者さんが二次治療のみ.39%の患者さんが三次治療以上の治療を受けていました。 白金製剤を含む化学療法への反応性は.完全寛解が47%.部分寛解が53%であり.ECOG fitness scoreは0が81%と大半の症例で認められた。 オラパリブ群とプラセボ群では.それぞれ17%と20%の患者さんがベバシズマブによる前治療を受けていました。
主要評価項目は無増悪生存期間(PFS)で.RECIST(Remission Evaluation Criteria in Solid Tumours)1.1評価法を用いて治験医師により測定されました。 副次的評価項目は.二次疾患進行または死亡までの期間(PFS2).OS(全生存期間).治療中止または死亡までの期間(TDT).最初の抗がん剤治療開始または死亡までの期間(TFST).二次抗がん剤治療開始または死亡までの期間(TSST).HRQoL(健康に関する生活の質)とし.これらの評価項目が達成されたことを確認した上で.本試験の結果を発表しました。 ).
本試験では.主要評価項目であるPFSがプラセボ群に対してオラパリブ群で統計的に有意に改善し.リスク比(HR)は0.30(95%CI 0.22-0.41; p<0.0001; 中央値はオラパリブ群19.1カ月に対してプラセボ群5.5カ月)であった。 治験責任医師が評価したPFSの結果は.盲検化された独立した中央画像評価によって裏付けられました(HR=0.25.95% CI 0.18-0.35.p<=0.0001.中央値はオラパリブ群30.2カ月.プラセボ群5.5カ月)。2年後.プラセボ群と比較してオラパリブ投与群の43%が無進行を維持しました。 は15%にとどまりました。
SOLO2におけるgBRCA1/2m PSR卵巣がん患者様の主要評価項目の結果概要については.表5および図1をご覧ください。
表5 SOLO2におけるgBRCA1/2m PSR 卵巣癌患者の主要評価項目。 試験結果の概要(治験責任医師による評価)。
| Olaparib300 mg錠。 (n=196) |
プラシーボ | |
| PFS(63%) 成熟度 | ||
| 107:196(55) | 80:99 (81) | |
| 期間中央値(月)(95%CI) | 19.1(16.3〜25.7) | 5.5(5.2-5.8) |
| HR (95% CI)a | 0.30 (0.22-0.41) | |
| P値(両側) | p<0.0001 | |
。
a HR=危険率。 Value<1 はオラパリブを支持する。 白金製剤を含む前治療の化学療法における寛解反応(CRまたはPR)および病勢進行までの期間(>6-12ヶ月および>12ヶ月)で層別化し.log-rank検定を用いて解析を行った。
bd 1日2回投与;PFS 無増悪生存期間;CI 信頼区間。
図1 SOLO2: gBRCA1/2m PSR 卵巣癌患者のPFSのKaplan-Meier曲線(63%)。 >成熟度 – 調査員評価)。
| ランダム化以降の期間(月) |
| プラシーボ |
| Olaparib300 mg 1日2回 |
| 危険にさらされている患者数 |
| プラシーボ |
| Olaparib300 mg 1日2回 |
bd 1日2回; PFS 無増悪生存期間 (n=99)
。 。 。 (n=10)
。 。 strong>日
。 。
。 。
。 bd>。
。 bd>。
。 。
。
。 。 BRCA1/2BRCA1/2
。
。 。 。
。 bd>。
。 bd>。
。
。
。
。 。
副次評価項目であるTFSTとPFS2は.プラセボ群と比較してオラパリブ群で持続的かつ統計的に有意な改善が認められました(表6)。
表6 SOLO2gBRCA1/2m PSR 患者における主要な二次試験について。 エンドポイント結果の概要
Olaparib300 mg錠。
(n=196) プラシーボ
TFST (58% maturity)
インシデント数:総患者数(%)
92:196 (47)
79:99(80)
期間中央値(月)(95%CI)
27.9(22.6-NR)
7.1(6.3-8.3)
HR(95%信頼区間)a
0.28(0.21-0.38)。
P値*(バイラテラル)
p<0.0001
PFS2(40%) 成熟度
インシデント数:総患者数(%)
70:196 (36)
49:99 (50)
期間中央値(月)(95%CI)
NR(24.1-NR)
18.4(15.4-22.8)
HR(95%信頼区間)a
0.50(0.34-0.72)。
P値(両側)
p=0.0002
* 制御不能な多重性
a HR=リスク比。 値 <1 はオラパリブを支持する。 白金製剤を含む前治療の化学療法における寛解反応(CRまたはPR)および病勢進行までの期間(6-12ヶ月および12ヶ月)で層別化し.log-rank検定を用いて解析した。
bd 1日2回投与.達成度はNR.CI信頼区間.PFS2は2回目の病勢進行または死亡までの時間.TFSTは最初の追跡治療開始または死亡までの時間。
本試験に登録された測定可能病変(ベースライン時の標的病変)を有する患者さんのうち.客観的寛解率は.プラセボ群17%に対し.オラパリブ錠群41%でありました。 試験開始時に病変(ベースライン時の標的病変または非標的病変)を有していたオラパリブ錠剤投与患者の15.0%で完全寛解が得られたのに対し.プラセボ投与患者では9.1%であった。
患者さんの自己報告による転帰(PRO)データでは.ベースラインの変化に対するFACT-O(Functional Assessment of Cancer Therapy-Ovarian Cancer)試験の転帰指標(TOI)で評価すると.オラパリブ群とプラセボ群との間に差は見られませんでした。
SOLO2研究 (D0816C00002)
研究 () 研究 () strong>–中国コーホート。
SOLO2試験には.中国人患者さんが別のコホートとして登録され.合計32名の中国人患者さんが.病勢進行または忍容できない毒性が発現するまで.300mg(150mg×2錠)を1日2回投与(n=22)またはプラセボ錠を1日2回投与(n=10)のいずれかにランダムに割り振られました。 中国人の被験者は深センのUW Clinical Laboratoryで検査を受けてgBRCA変異があることが確認された。 本製品を投与された患者さんの年齢中央値は49歳(範囲:37~65歳).プラセボを投与された患者さんの年齢中央値は46.5歳(範囲:33~67歳)となっています。 ECOGフィジカル・ステータス・スコアは.治療群で73%.プラセボ群で50%が0でした。全患者の59%が無作為化前の最後の白金製剤を含む化学療法後に完全寛解を達成し.56%の患者は最後の白金製剤を含む化学療法完了後6〜12カ月で病勢進行までの時間を達成しました。 オラパリブ群の約23%.プラセボ群の約20%が.3ライン以上の白金製剤を含む前治療を受けていました。 SOLO2 Chinaのコホートにおいて.本製品はプラセボと比較して.患者さんのPFS(治験担当医師による評価)をHR 0.44(95%CI:0.17-1.19.p=0.0776.中央値:オラパリブ群13.8ヶ月.プラセボ群5.5ヶ月)と.有意に改善させました。 この結果は.独自に行った画像評価の結果とも一致します。 患者の生存率に関するデータはまだ成熟していない(19%の患者にのみイベントが発生した)。
表7 SOLO2Chinese CohortgBRCA1/2mPSR 卵巣癌の患者さん 主要評価項目(治験責任医師評価)の結果のまとめ。
オラパリブ錠 300 mg 錠 1日2回投与!
(n=22)プラシーボ
PFS(65.6%) 成熟度
イベント数:総患者数(%)
14:22 (63.6)
7分10秒(70.0)
期間中央値(月)(95%CI)
13.8 (10.1, 16.6)
5.5(3.2.8.4)
リスク比 HR (95% CI)a
0.44 (0.17, 1.19)
P値(bilateral)a
p=0.0776
a HR=リスク比.リスク比対p値は非階層化比例リスクモデルによるもの。
PFS 無増悪生存期間
中国人を対象とした有効性および安全性は.非中国人を対象とした有効性および安全性と同様であった。
Study19(D0810C00019Study 19 em>)。
19試験は.プラチナ製剤による2種類以上のレジメン投与後のPSR卵巣がん患者(卵管がん.原発性腹膜がんを含む)を対象に.維持療法としてのオラパリブの安全性と有効性を評価する無作為化二重盲検プラセボ対照の第II相試験です。 白金製剤を含む化学療法終了後に寛解(CRまたはPR)を得たPSRグレードのプラズマ卵巣がん患者265名を登録し.1:1で無作為化(オラパリブ群136名.プラセボ群129名)し.疾患進行または忍容できない毒性が発現するまでオラパリブのカプセル400mg(50mg×8錠)1日2回(中国ではカプセルの入手は申告されていません)とプラセボの投与を実施しました。 主要評価項目はRECIST 1.0基準を用いて治験責任医師が評価したPFSで.副次的評価項目はOS.疾患制御率(DCR.すなわちCR/PR+SD(疾患安定化)確認).HRQoL.疾患関連症状などでした。 TFSTとTSSTについては.探索的な解析を行った。
本試験では.白金製剤を含む最後の化学療法終了から疾患再発までの6ヶ月間の患者を登録しました。 組み入れ条件には.BRCA1/2変異の同定は含まれなかった(一部の患者はBRCA変異の状態についてレトロスペクティブに検査されている)。 オラパリブまたは他のPARP阻害剤による前治療を受けていない患者であること。 ベバシズマブによる治療歴があってもよいが.無作為化前の治療レジメンにベバシズマブが含まれていないことが条件とされた。 オラパリブ治療中に病勢進行が生じた場合.それ以降のオラパリブによる治療はできません。
BRCA1/2変異を有する患者は.局所検査または血液生殖細胞検査用のMyriad CLIA comprehensive BRACAnalysis® 検査.あるいはFoundation Medicineが実施する検査を用いて腫瘍サンプルで行われる検査を用いて同定されました。 無作為化された患者の7.4%(10/136人)でBRCA1/2遺伝子にラージフラグメント再配列が検出された。
人口統計学的特性およびベースライン特性は.オラパリブ群とプラセボ群で基本的に類似していました。 両群の年齢の中央値は59歳で.86%が原発性卵巣癌であった。 オラパリブ群では.過去に二次治療のみを受けていた患者さんが44%.三次治療以上を受けていた患者さんが56%でした。 プラセボ群では.49%の患者さんが二次治療のみ.51%の患者さんが三次治療以上の治療を受けていました。 白金製剤を含む化学療法に反応した患者は.45%が完全寛解.55%が部分寛解であった。 オラパリブ群およびプラセボ群では.ベバシズマブによる前治療を受けた患者はそれぞれ6%と5%でした。
本試験では.主要評価項目であるPFSがプラセボ群に比べオラパリブ群で統計的に有意に改善し.HR 0.35 (95% CI 0.25-0.49; p<0.00001; 中央値はオラパリブ群 8.4 ヶ月 対 プラセボ群 4.8 ヶ月)となりました。 最終的なOS解析(成熟度79%.データカットオフ日[DCO]2016/05/09)では.オラパリブとプラセボのリスク比は0.73(95%CI 0.55-0.95;p=0.02138[specified significance level not reached<;0.0095]; 中央値はオラパリブ群では29.8ヶ月.プラセボ群では (27.8ヵ月)。 オラパリブ投与群では.23.5%(n=32/136)の患者が2年以上の治療を受けたのに対し.プラセボ投与群では3.9%(n=5/128)の患者が2年以上の治療を受けていました。 患者数が限られているにもかかわらず.オラパリブ群の13.2%(n=18/136)が5年以上治療を受けたのに対し.プラセボ群では0.8%(n=1/128)でした。
事前に計画されたサブグループ解析の結果.卵巣がんのBRCA1/2変異を有する患者さん(n=136, 51.3%; 体細胞BRCA1/2変異が確認された20名を含む)には.olaparib単独療法の維持療法の効果が最も高いことがわかりました。 また.BRCA1/2野生型/意義不明の変異体(BRCA1/2 wt/VUS) 患者においても.より少ないながらも臨床的有用性が観察されました。 サブグループ解析のための多重検定ができなかった。
19試験におけるBRCA1/2変異およびBRCA1/2 wt/VUS PSR卵巣がん患者における主要試験評価項目の結果の概要は.表8と図2をご覧ください。
表8 研究 19BRCA1/2
strong>変異とBRCA1/2 wt/VUS PSR 卵巣癌患者における主要評価項目の結果概要
卵巣癌患者における主要評価項目の結果は.次のとおりです。
All patientsa
BRCA1/2mutations
BRCA1/2 WT/VUS
Olaparib400 mg.
カプセルbdプラシーボ
Olaparib400 mg.
カプセルbdプラシーボ
Olaparib400 mg.
カプセルbdプラシーボ
PFS-DCO 2010年06月 302010
イベント数:総患者数
(%)60:136
(44)94:129
(73)26:74
(35)46:62
(74)32:57
(56)44:61
(72)
期間中央値(月)(95%CI)
8.4
(7.4-11.5)4.8
(4.0-5.5)11.2
(8.3-NR)4.3
(3.0-5.4)7.4
(5.5-10.3)5.5
(3.7-5.6)
HR (95% CI)b
0.35(0.25〜0.49)
0.18(0.10〜0.31)
0.54(0.34〜0.85)
P値(両側)
p<0.00001
p<0.00001
p=0.00745
a 以下のサブグループを含む全患者: BRCA1/2-mutation, BRCA1/2 wt/VUS and BRCA1/2 status unknown (11 patients with unknown states not included in the
表は.独立したサブグループとしてではなく)。
b HR=リスク比。 値 <1 はオラパリブを支持する。 解析は.治療法.民族的起源.プラチナ感受性についてCox比例リスクモデルを用いて行われた。
と最後の白金製剤ベースの化学療法後の寛解をエフェクターとして使用した。
wt(野生型)野生型;VUS(意義不明の変異)意義不明の変異;bd 1日2回;PFS 無増悪生存期間;DCOデータカットオフ日;CI 信頼区間;NR 未達成。
図219試験:FASにおけるPFSのKaplan Meier カーブ(58%):FASにおけるPFS カーブ。 strong>Mature –Researcher Assessment) DCO 2010 06 30 6月 Mature –Multi –Multi –
ランダム化以降の期間(月)
プラシーボ
Olaparib400mg
Olaparib400mg
プラシーボ
危険にさらされている患者数:
bd 1日2回; DCO data cut-off date; FAS full analysis set; PFS progression-free survival
19試験におけるBRCA1/2変異およびBRCA1/2 wt/VUS PSR卵巣がん患者さんの主要副次評価項目の結果を表9に.全患者さんの結果を図3にまとめました。
表9 研究 19BRCA1 >/2変異とBRCA1/2 wt/VUS PSR 卵巣癌患者における主要副次評価項目の結果概要 卵巣癌患者における主要副次評価項目の結果概要。 強い>。
All patientsa
BRCA1/2mutations
BRCA1/2 wt/VUS
Olaparib400 mg.
カプセルbdプラシーボ
Olaparib400 mg.
カプセルbdプラシーボ
Olaparib400 mg.
カプセルbdプラシーボ
OS – DCO 2016Year05Month09Year05Month strong>日
インシデント数:総患者数(%)
98:136
(72)112:129
(87)49:74
(66)50:62
(81)c45:57
(79)57:61
(93)
平均時間(月)
(95% CI)29.8
(26.9-35.7)27.8
(24.9-33.7)34.9
(29.2-54.6)30.2
(23.1-40.7)24.5
(19.8-35.0)26.6
(23.1-32.5)
HR (95% CI)b
0.73(0.55〜0.95)
0.62(0.42〜0.93)
0.84(0.57〜1.25)
P値*(バイラテラル)
p=0.02138
p=0.02140
p=0.39749
TFST – DCO 2016Year05Month09Year05Month0strong>Year/No. strong>日
インシデント数:総患者数(%)
106:136
(78)124:128
(97)55:74
(74)59:62
(95)47分57秒
(83)60:61
(98)
平均時間(月)
(95% CI)13.3
(11.3-15.7)6.7
(5.7~8.2)15.6
(11.9-28.2)6.2
(5.3-9.2)12.9
(7.8-15.3)6.9
(5.7-9.3)
HR (95% CI)b
0.39(0.30〜0.52)
0.33(0.22〜0.49)
0.45(0.30〜0.66)
P値*(バイラテラル)
p<0.00001
p<0.00001
p=0.00006
* サブグループ解析や全患者のTFSTに多重検定戦略はなかった。
aすべての患者さんには以下のサブグループが含まれます:BRCA1/2変異.BRCA1/2 wt/VUS および BRCA1/2 状態不明(11名は.BRCA1の状態不明)。
表は.独立したサブグループとしてではなく)。
b HR=リスク比。 値 <1 はオラパリブを支持する。 解析は.Cox比例リスクモデルを用いて.治療法.人種.プラチナ感受性.および
白金製剤を用いた最終化学療法後の寛解をエフェクターとする。
cBRCA変異サブグループでは.プラセボ投与群の約4分の1(14/62;22.6%)が.その後PARP阻害剤による治療を受けました。
wt(野生型)野生型;VUS(意義不明の変異)意義不明の変異;bd 1日2回;OS 全生存期間;DCO データカットオフ;CI:信頼区間;TFST ランダム化から最初の後続治療または死亡までの時間
<図3 試験19:FASにおけるOS(79%)のカプラン・マイヤー曲線。 strong>Maturity, DCO 2016 May 09) Maturity, DCO 2016
プラシーボ
Olaparib400mg
Olaparib400mg
Placebo
危険にさらされている患者数:
ランダム化以降の期間(月)
bd 1日2回; DCO data cut-off date; FAS full analysis set; OS overall survival
患者さんの自己報告による転帰(PRO)データでは.Functional Assessment of Cancer Therapy – Total Ovarian Cancer Score(FACT-O)のTrial Outcome Index(TOI)で測定した改善率および悪化率により.olaparib群とプラセボ群に差がないことを示しました。
【薬理学・毒性学】
薬理作用。
Olaparibは.ポリADPリボースポリメラーゼ(PARP.PARP1.PARP2.PARP3)阻害剤であり.PARPは.DNA転写やDNA修復などの正常な細胞機能に関与している。 オラパリブは.in vitroでは腫瘍細胞株の増殖を.in vivoではヒト腫瘍搭載マウス異種移植片の増殖を抑制し.単剤またはプラチナ製剤による化学療法後の投与で有効であることが試験で示されています。 オラパリブ投与により.白金系化学療法反応に伴うDNA損傷相同組換え修復のBRCA関連欠損.または非BRCA関連欠損を有する細胞株およびマウス移植腫瘍モデルにおいて細胞障害および腫瘍抑制作用が増強されました。 In vitroの研究では.オラパリブの細胞毒性は.PARPase活性の阻害とPARP-DNA複合体形成の増大が関与し.DNA損傷とがん細胞死を引き起こすことが示唆されています。
毒性試験。
遺伝毒性: オラパリブのエームス試験の結果は陰性でした。中国ハムスター卵巣(CHO)細胞染色体異常試験およびラット骨髄小核試験の結果は染色体切断作用が陽性で.オラパリブの薬理作用と一致しました。 ゲノムの不安定性はオラパリブの薬理作用と一致し.オラパリブがヒトにおいて遺伝毒性を有する可能性が示唆された。
<生殖毒性:雌ラットの生殖能力及び初期胚発生毒性試験において.交配14日前から妊娠6日目までオラパリブを最大15mg/kg/日(母親の全身曝露量は臨床推奨ヒト曝露量[AUC0-24h]の約7%)で経口投与したところ.以下の影響は認められませんでした。 交配および生殖能力への影響は見られなかったが.生殖後の流産が増加した。
雄ラットの生殖能力試験において.オラパリブを40mg/kg/日までの用量(全身曝露量は臨床推奨ヒト曝露量[AUC0-24h]の約5%)で少なくとも70日間経口投与したところ.交尾及び生殖能力に対する影響は認められなかった。
胚・胎児発生毒性試験において.器官形成期の妊娠ラットにオラパリブ 0.05 および 0.5 mg/kg/day を経口投与した。 胚・胎児毒性は0.5 mg/kg/日の用量(母親の全身曝露量はヒトの臨床推奨曝露量[AUC0-24h]の約0.18%)で見られ.到着後の流産の増加.眼の融合または欠損(無眼症.小眼症).脊椎・あばら(あばらまたは骨化中心の追加.脊椎弓.あばらおよび胸骨).頭蓋骨(後頭部融合)および横隔膜(ヘルニア)が含まれていた。 外固定).重度の横隔膜の奇形(ヘルニア)などがあります。 その他.骨化不全や骨化欠如(椎骨/胸骨.肋骨.四肢).椎骨/胸骨.骨盤帯.肺.胸腺.肝臓.尿管.臍動脈などの異常や変種があります。 0.05mg/kg/dayの投与量では.上記の眼球.肋骨および尿管の異常の発生率は低いものでした。
発がん性: 関連する試験は実施されていない。
【ファーマコキネティクス】
オラパリブの剤形は.錠剤とカプセル剤(カプセル剤は中国では未申告)です。 錠剤の経口バイオアベイラビリティは.カプセルのそれよりも高いです。 母集団薬物動態解析の結果.300mg錠剤を1日2回投与した場合の定常状態の曝露量(AUC)は.400mgカプセルを1日2回投与した場合よりも77%高いことが示された。300mg錠剤単回投与後のオラパリブのAUCおよびCmaxの幾何平均値は42.0 μg*h/mL(n=204) および 5.8 μg/mL(n=204) であった。204)であり,300 mg錠を1日2回投与したときの定常状態のAUCおよびCmaxの幾何平均値はそれぞれ49.0 μg*h/mL(n = 227)および7.7 μg/mL(n = 227)であった。 オラパリブのPKは時間依存的であり.複数回の投与により定常状態のクリアランスが15%減少した。
吸収。
オラパリブの経口投与では.吸収が速く.血漿中のピーク濃度の中央値は通常投与後1.5時間で到達します。300mg錠を1日2回反復投与した場合.定常状態のAUC平均集積比は1.8となりました。
オラパリブの全身曝露量(単回投与AUC)は.用量範囲が25mgから450mgの場合.用量にほぼ比例して増加し.Cmaxは同じ用量範囲では用量増加にやや比例せず増加しました。
高脂肪食との併用により.オラパリブの吸収速度(tmaxが2.5時間遅延)が増加したが.オラパリブの吸収範囲に大きな変化はなかった(平均AUCが約8%増加)。
ディストリビューション。
オラパリブ300mg単回投与後のオラパリブの平均(±標準偏差)見かけの分布容積は158±136Lであり.in vitroでの蛋白結合率は約82%であった。
メタボリズム。
in vitro試験において.CYP3A4/5はオラパリブの代謝に主に関与する酵素であることが示されています。
女性患者では.プロトタイプのオラパリブを経口投与した後.血漿中に循環する放射能の大部分(70%)が14C-オラパリブで占められています。 14C-オラパリブは広範囲に代謝され.プロトタイプ薬剤は尿中で15%.糞便中で6%を占めています。 代謝のほとんどは酸化反応に起因し.生成された成分の多くは.その後グルコシノレートや硫酸塩の結合を受ける。
排他的。
オラパリブ300 mg単回投与時の平均血漿終末半減期(±標準偏差)は14.9 ± 8.2時間.見かけの血漿クリアランスは7.4 ± 3.9 L/hであった。
14C-olaparib単回投与後.与えられた放射能の86%が7日間の収集期間内に回収され.44%が尿から.42%が糞便から回収されました。 大半は代謝物として排泄された。
特殊な人口。
集団薬物動態解析において.患者の年齢.性別.体重.人種(白人.中国人.日本人を含む)は.有意な共変量ではありませんでした。
肝機能障害肝機能障害の試験では.軽度の肝機能障害患者(Child-Pugh分類A;n=9)にオラパリブを投与した場合.肝機能正常患者(n=13)と比較して.平均AUCが15%.平均Cmaxが13%増加しました。 軽度の肝障害はオラパリブのタンパク質結合に影響を及ぼさないため.血漿中の総露出量は遊離した薬剤を表しています。 中等度または重度の肝障害を有する患者におけるデータは入手できない。
腎臓障害。
腎機能障害に関する試験では.腎機能正常者(CLcr≧81mL/min;n=12)と比較して.オラパリブを服用した軽度腎機能障害者(Cockcroft-Gault式で定義.CLcr=51~80mL/min;n=13)は.平均AUC増加率が24%.平均Cmax0… sub>が15%増加し.中等度の腎障害患者(CLcr = 31-50 mL/min.n=13)ではオラパリブの服用によりAUCとCmaxの平均値がそれぞれ44%と26%増加しました。 オラパリブの血漿蛋白結合の程度とクレアチニンクリアランスの間に相関を示す証拠はなかった。 重度の腎障害または末期腎不全(CLcr≦30mL/min)の患者に関するデータはありません。
【ストレージ】。
30℃以下で保存してください。
【パッケージ】。
アルミニウム-アルミニウムブリスターパック.1箱56錠入り(7プレート)
アルミニウム-アルミニウムブリスターパック.1箱112錠入り(14枚入り)
[有効期限]。
36ヶ月
【エグゼクティブ・スタンダード】
輸入医薬品の登録に関する基準。
【承認番号】
会社名:AbbVie Deutschland GmbH & Co. KG
生産拠点住所:Knollstrasse 67061 Ludwigshafen, Germany
中国駐在員事務所住所:江蘇省無錫市新区黄山路2号
郵便番号:214028
品質に関する苦情:400 828 1755.800 828 1755
製品情報無償提供:400 820 8116, 800 820 8116
ファックス: 021-38723255
ウェブサイト:www.astrazeneca.com.cn.